Professional Documents
Culture Documents
Enzyme Biotechnology by Bhatt Amity HTTP Process Technology Wet Paint Com
Uploaded by
raogh77Original Description:
Original Title
Copyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
Enzyme Biotechnology by Bhatt Amity HTTP Process Technology Wet Paint Com
Uploaded by
raogh77Copyright:
Available Formats
Dr. S. M. Bhatt smbhatt@amity.edu , smbhatt_bhu@rediffmail.
com phone 9313993840 Amity University Uttar Pradesh ENZYMOLOGY AND ENZYME TECHNOLOGY Course Code: BTBBT 30602
Course Objective: The course aims to provide an understanding of the principles and application of proteins, secondary metabolites and enzyme biochemistry in therapeutic applications and clinical diagnosis. The theoretical understanding of biochemical systems would certainly help to interpret the results of laboratory experiments. Course Contents: Module I Enzymes: Introduction and scope, Nomenclature, Mechanism of Catalysis. Module II Specificity of enzyme action, monomeric and oligomeric enzymes, Enzyme inhibition. Module III Enzyme Kinetics: Single substrate steady state kinetics; Michaelis Menten equation, Linear plots, King-Altmans method; Inhibitors and activators; Multisubstrate systems; ping-pong mechanism, Alberty equation, Sigmoidal kinetics and Allosteric enzymes Module IV Extraction & purification of enzymes. Module V Immobilization of Enzymes; Advantages, Carriers, adsorption, covalent coupling, cross-linking and entrapment methods, Micro-environmental effects. Module VI Biotechnological applications of enzymes: Large scale production and purification of enzymes, enzyme utilization in industry, enzymes and recombinant DNA technology Examination Scheme: Component Codes Weight age (%) H/Q 10 S 10 CT2 20 EE 60
Text & References: Text Biotechnological Innovations in Chemical Synthesis, R.C.B. Currell, V.D. Mieras, Biotol Partners Staff, Butterworth Heinemann. Enzyme Technology, M.F. Chaplin and C. Bucke, Cambridge University Press. Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis, R.A. Copeland, John Wiley and Sons Inc. References Enzymes Biochemistry, Biotechnology, Clinical Chemistry, Trevor Palner Dr. S. M. Bhatt smbhatt@amity.edu , smbhatt_bhu@rediffmail.com phone 9313993840 Amity University Uttar Pradesh Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems, I.H. Segel, Wiley-Interscience
Industrial Enzymes & their applications, H. Uhlig, John Wiley and Sons Inc.
ENZYMOLOGY AND ENZYME TECHNOLOGY LAB Course Code: BTBPL 30622
Course Objective: The laboratory will help the students to isolate enzymes from different sources, enzyme assays and studying their kinetic parameters which have immense importance in industrial processes. Course Contents: Module I Isolation of enzymes from plant and microbial sources. Module II Enzyme assay; activity and specific activity determination of amylase, nitrate reductase, cellulase, protease Module III Purification of Enzyme by ammonium sulphate fractionation Module IV Enzyme Kinetics: Effect of varying substrate concentration on enzyme activity, determination of Michaelis-Menten constant (Km) and Maximum Velocity (Vmax.) using Lineweaver-Burk plot. Module V Effect of Temperature and pH on enzyme activity Module VI Enzyme immobilization Examination Scheme: Major Experiments: Minor Experiments: Spotting: Viva: Records: Total: 40 20 10 100
10 20
Note: Minor variation could be there depending on the examiner. Text: Practical Biochemistry, Sawhney and Singh
Chapter- 1
Enzymes an introduction 9
Definition Introduction
Historical aspects in enzyme discovery Classification of Enzymes. application of enzymes Chapter-2 Types of Enzymes 16 A. Simple enzymes B. Complex enzymes Complex enzymes Role of Coenzymes A. Catalysts B. Biocatalyst or Enzymes How enzymes speed up the reaction without taking part Unit of activity Chapter 3 Enzyme Specificity 20 ENZYME MECHANISM MODELS A. Lock and Key Model for Enzyme Activity B. Induced Fit (Hand and Glove) Model of Enzyme Activity Mechanisms of Enzyme Catalysis Chapter 4 Enzyme-Substrate Interactions 24 a. Catalysis by Bond Strain b. Catalysis by Proximity and Orientation c. Catalysis Involving Proton Donors (Acids) and Acceptors (Bases) Concerted acid-base catalysis d. Covalent Catalysis: Covalent Intermediates e) Metal Ion Catalysis Catalytic Power a) Proximity b) Orientation f. Catalysis in organic solvents Some advantages to using enzymes in non-aqueous solvents include (Dordick, 1989): Importance and relevance of whole cell enzymes in biotechnology There may be some drawbacks to using whole cells in catalysis. These include Abzymes Exercises Chapter-5 ENZYME KINETICS & INHIBITION 46 Chemical Reactions and Rates Chemical Reaction Order Michaelis-Menton constant a. Pre-steady state:-b. Steady state:-Derivation: Catalytic efficiency and Turnover number. Chapter-6 Factor affecting the rate of catalysis 58 Substrate concentration Effect of temperature and pressure Effect of pH on Enzyme Chapter-7 Enzyme inhibition 68 A. Competitive inhibition B. Uncompetitive inhibition C. Noncompetitive inhibition Determination of Vmax and Km Chapter-8 Multisubstrate enzymes 83 Enzyme Catalysis
Mechanisms of Chemical Catalysis one-substrate enzymes: Linear plots for enzyme kinetic studies FYI - The Eadie-Hofstee Plot ENZYME KINETICS AND INHIBITION What's exciting about enzyme inhibition? Multi-substrate Enzymes Chapter-9 ISOLATION of ENZYME 102 INTRODUCTION strategies for enzyme purification. Method of separation. Test of purity is done by following method Source of enzyme Media for enzyme production Cell can be break down byfollowing methods A. Cell breakage by osmotic shock B. Use of enzymic lytic methods c. Ultrasonic cell disruption d. Cell lysis by High pressure homogenisers Method of separation of enzyme from lysed cell; CentrifugationB. ULTRACENTRIFUGATION A. Differential centrifugation Filtration of enzyme after separation Separation of enzyme in Aqueous biphasic systems Preparation of enzymes from clarified solution; Ultrafiltration Chapter-10 Concentration and purification of enzyme 123 Concentration by precipitation A. Nucleic acid removal B. Heat treatment 1. Enzyme purification by Chromatography 2 Affinity chromatography .3 Hydrophobic interaction chromatography (HIC) or affinity elution CHOICE OF MATRIX Electrophoresis 2.11 Maintaining Enzyme Activity Chapter-11 Techniques used in Enzyme characterization 132 HOW TO KNOW THE PROPERTY OF ENZYME Introduction A. Chromatography Principles of chromatography Adsorption Chromatography COLUMN CHROMATOGRAPHY History Principle Mechanism of separation by size in gel permeation chromatography ADVANTAGE OF GEL CHROMATOGRAPHY Application of GPC or gel permeation chromatography PAPER CHROMATOGRAPHY Ion exchange chromatography There are two type of ion exchanger Choosing an Ion Exchanger Gas / Liquid Chromatography Reversed Phase Chromatography HIGH PRESSURE LIQUID CHROMATOGRAPHY [HPLC] Partition Chromatography
127
Affinity chromatography Principle METHOD OF LIGAND IMMOBILIZATION 1. CNBR activated agarose 2. 6-AMINO-HEXENOIC ACID & 1,6 DIAMINO HEXANE. 3. Carbomyl diimidazole activated agarose. Applications Thin layer chromatography Principle ADVANTAGE OF TLC OVER OTHER CHROMATOGRAPHY TECHNIQUE HPTLC and HPPLC Selection of chromatographic system Electrophoretic technique a. Agarose Gel Electrophoresis b. Polyacrylamide Gel Electrophoresis c. SDS-PAGE f. Iso-electric focusing: IEF gel: g. Chromatofocussing 2D-PAGE DETECTION, ESTIMATION AND RECOVERY OF PROTEIN GELS. i. AGAROSE GEL ELECTROPHORESIS OF DNA Preparation of the gel Polyacrylamide gel Chapter -12 Enzyme Immobilization 160 Introduction Aim of Enzyme Immobilization [a] Physical properties [b] Chemical properties [c] Stability [d] Resistance [e] Safety [f] Economic Limitations of Immobilized Enzyme Advantages of Immobilizations Methods of immobilizations Carrier matrices Adsorption of enzymes Some disadvantages of adsorption include Covalent coupling Functional groups that affects the covalent coupling A. cyanogen bromide (b) Ethyl chloroformate (c) Carbodiimide (d) Glutaraldehyde (e) 3-aminopropyltriethoxysilane Entrapment and Encapsulation Entrapment of enzymes Membrane confinement Encapsulation of Enzymes Crosslinking Chapter-13 Introduction to Industrial biotechnology 171 Application of Immobilised-Enzyme Processes Introduction 172 1-High-fructose corn syrups (HFCS) 2-GLUCOSE ISOMERASE a Treatment with activated carbon.
3-Use of immobilised raffinase 4-Use of immobilised Invertase 5-Production of amino acids 6- Use of immobilised lactase .7 Production of antibiotics Preparation of acrylamide Chapter-14 Application of Enzyme in clinics 233 Introduction Determination of enzyme activities 1- Clinical Enzymology of liver disease. 2- In Heart disease -amylase Creatine Kinase and fructose bisphosphate aldolase. Alkaline phosphates Acid phosphatase. In Treatment of cancer Enzyme deficiencies Enzyme inhibitors and drug design 183 Use of enzyme in Dignosis Blood glucose Disadvantages of Using Enzyme as Therapeutic Agents Thermozymes The large-scale use of enzymes in solution The use of enzymes in detergents Chapter-15Application of Enzyme in Industries different sector of industry where enzymes are used Food Textile and other indsutries Chapter-16 Enzyme Reactors all type of reactors detailed dscription kinetics involved in reactors Chapter-17 Protein Engineering 215 Artificial enzymes Chapter-18 Biosensors and Immunosensors 225 Chapter-19 Enzyme and protein STABILITY 246 I Definition of Stability II The Unfolded State III Major Factors Affecting Protein Stability The Hydrophobic Effect Hydrogen Bonds Conformational Entropy of Unfolding IV Other Factors Affecting Protein Stability Salt Bridges Aromatic-Aromatic Interactions Metal Binding Disulphide Bonds V Chemical Degradation VI Conclusions Chapter -20 Study of drug and molecule binding stratgies 260 TYPES OF DNA BINDING DRUGS NON COVALENT INTERACTIONS Minor Groove binders INTERCALATORS COVALENT INTERACTIONS DNA-Drug Interaction : Chapter-21 Technique to study protein 284
Nuclear Magnetic Resonance(NMR)Spectroscopy Determination of membrane protein structure by rotational resonance NMR: bacteriorhodopsin. Structure of bacteriorhodopsin: Determination of membrane protein structure by rotational resonance NMR: bacteriorhodopsin Chapter-22 Enzymology of DNA folding and unfolding 290 WHAT IS DNA Major and minor grooves Replication Enzyme involved in DNA folding and unfolding Invitro-DNA folding and unfolding Folding unfolding kinetics Chapter-23 Safety and regulatory aspects of enzyme use 305
Module I Enzymes: Introduction and scope, Nomenclature, Mechanism of Catalysis. Chapter- 1 Enzymes an introduction Definition Calorimetry: Measurement of heat released or taken up in a chemical reaction or physical process to derive thermodynamic data (e.g., dissociation constant, freeenergy change, enthalpy change, entropy change) for molecular interactions. Two kinds of calorimetry important to biomolecular studies are isothermal titration calorimetry (ITC), which can be used to detect the number of binding sites on an enzyme; and differential scanning calorimetry (DSC), which monitors. Enzyme: Protein that acts as a catalyst, speeding the rate of a biochemical reaction but not altering its direction or nature. Electrophoresis: Method of separating large molecules (such as DNA fragments or proteins) in a sample. An electric current is passed through a medium containing the sample; each molecule travels through the medium at a different rate, depending on its electrical charge, shape and size. Agarose and acrylamide gels are commonly used media for electrophoresis of proteins and nucleic acids. In onedimensional (1D) gel electrophoresis, proteins and nucleic acids are separated in one direction on a gel, primarily by size. Two-dimensional (2D) gel electrophoresis is used in proteome analyses to separate complex protein mixtures using two separation planes (e.g., vertically down a gel by net charge and horizontally by molecular mass). Each unique protein mixture produces a characteristic pattern or fingerprint of protein separation on a 2D gel.
Flow Cytometry: Analysis of biological material by detection of light-absorbing or fluorescing properties of cells or subcellular components (e.g., chromosomes) passing in a narrow stream through a laser beam. An absorbance or fluorescence profile of the sample is produced. Automated sorting devices, used to fractionate samples, separate successive droplets of the analyzed stream into different fractions depending on the fluorescence emitted by each droplet. Kinetics: Field of study that deals with determining the rates of biological, chemical, and physical processes (e.g., how quickly reactants are converted into products) under various conditions. Metalloprotein: Protein that incorporates one or more metals into its molecular structure by binding individual metal ions [e.g., iron (Fe2+ or Fe3+), zinc (Zn2+), or magnesium (Mg2+)] or nonprotein organic compounds containing metals. Metalloprotein are important components of electron transport chains. Steady State: Growth state in which the concentration of bacterial cells is in equilibrium with the concentration of nutrients or substrates (i.e., the concentrations remain constant over time). X-ray crystallography: Technique used to obtain structural information for a substance (e.g., protein, molecular complex) that has been crystallized. A beam of X rays is focused on the crystals, and the scattering pattern of the X rays is used to create 3D representations of the crystal with atomic resolution.
Introduction Enzymes are the wonderful creations of the biological system. Various biological reactions go on smoothly without single mistakes. Enzymes have specificity not only toward the substrate but also recovered in the end of the reactions. They are also regulated very precisely. Normal monomeric enzyme like trypsin and pepsin are regulated by removing or adding a small piece of peptide that make them inactive or active while the complex enzymes are regulated by end product , or by allosteric mechanism. Homones in several cases also play important role in regulations like in formation of lactose by progestron and prolactin before and after the birth of baby. The system (complex enzymes are termed as system) specifically and precisely binds to the substrate and convert them into the required product that sometimes becomes the substrate for next reaction for next enzyme to act on. Total activity of enzyme depends on several factors like acid base, structural orientations and strain, electrostatic interactions (by metal ions) and formation of covalent bond. In several cases certain organic molecule or metals termed as co-enzyme or cofactor that either or electron or proton acceptor or by electrostatic mechanism are needed to convert a substrate to product. Actually enzymes activity depends on the precise function of a small part called as the active site that is a small pocket like structure that contains various helper amino acid residues that by electron or proton transport make bond of the substrate molecule weaker and rearrange them into a stable product at normal temperature and pressure. Any deficiency in the formation of these pockets like structure may render the whole enzyme to become inactive that leads to certain deficiency diseases. Modern techniques like MOLDI-TOF, x-ray crystallography along with sequencer helped a lot to scientist in deciphering the mechanism of enzyme activity. Knowledge of enzyme activity and its detailed mechanism made it possible to design artificial enzyme in the lab to cop up with many enzyme related metabolic problems or complex viral enzyme like AIDS. Historical aspects in enzyme discovery 1860 Bertholate disrupted yeast cell and isolated extract that catalyzed
conversion of 1878 Word enzyme first used by kuhne. 1894
sucrose to glucose and fructose.
Fisher proposed lock and key hypothesis to explain the mechanism of Enzyme operating between substrate and enzyme.
1897 Buchner isolated yeast extract used for formation of ethanol. 1920 Sumner crystallized Urease first time that hydrolyze the urea into CO2 and NH3. 1958 and 1960 1961 1965 1986 Koshland proposed induced fit model to account for enzyme catalytic power specificity. Ribonuclease amino acid was sequenced. Ribonuclease was artificially synthesized from amino acid precursor. three dimensional structure of Lysozyme was deduced. RNase was found to work as catalyst by Cech. They were called as ribozymes. e.g. Ribonuclease P Classification of Enzymes. The I.U.B. system also specifies a textual name for each enzyme. The enzyme's name is comprised of the names of the substrate(s), the product(s) and the enzyme's functional class. Because many enzymes, such as alcohol dehydrogenase, are widely known in the scientific community by their common names, the change to I.U.B.approved nomenclature has been slow. In everyday usage, most enzymes are still called by their common name. In 1955 International Union of Biochemistry classified the enzyme on the basis of kind of reaction they performed, substrate they act, position of group or functional group and number of amino acid. Therefore they divided the enzyme into six major classes. Suffix added in the enzyme is ase to the name of substrate or to word. Urea hydrolysis is done by the Urease. There are major 6 classes and each with the subclass. Each enzyme has assigned 4 digit classification number and a systematic name. For e.g. EC 2.7.1.1 means first digit indicates the class transferases, and the second digit denotes the subclass, third and four digits is the group transfer or accepted as shown in figure. Classes of enzyme: there are six classes of enzyme according to reaction type they performed 1. Oxidoreductases- These are enzymes that catalyze oxidations or reductions. Oxidation means transfer of oxygen atom and reduction means transfer of H atom. Enzymes such as dehyrogenases, oxidases, and peroxidases. Its second digit has been denoted in the table. Its third digit refers to the hydrogen or electron acceptor. For example 1-for NAD+ or NADP+ 2. For Fe+3 3. For oxygen. For clear understanding we can take example of S-latate NAD+ oxidoreductase (E.C. 1.1.1.27). Here S lactate is the substrate and NAD+ is electron acceptor/ hydrogen acceptor and the reaction is of type oxidation reaction. In EC number 1 digit denotes type of reaction i.e. oxidoreductase 2nd digit is the functional group of lactate which is alcohol therefore it is denoted by subclass 1, 3rd digit denotes NAD as electron acceptor NAD+, while 4th digit is the actual substrate.
Lactate dehydrogenase CH3CH(OH)COO- + NAD+ ---------- CH3C(O)COO- + NADH+ + H+ 2. Transferases- These enzymes catalyze the transfer of a group from one molecule to another. Examples such as Phosphatases, transaminases, and transmethylases. Its second digit has been provided in the table. Its third digit denotes the group transferred. Thus 1 denotes transfer of CH3 group while 2 denotes the transfer of CH2OH and 3 denotes the carboxyl or carbamoyl transferases. Phosphotransferases are given trivial name as kinase for example hexokinases (E.C. 2.7.1.1 or D-hexose-6-phosphotransferase, 7 for phosphate group, 1 for single carbon unit) that catalyses the transfer of phosphate from ATP to D hexose resulting the D hexose-6-phosphate. D-hexose-6-phosphotransferase (hexokinase) D hexose + ATP-----------------------------------phosphate + ADP D hexose-6-
3. Hydrolases-These enzymes catalyze hydrolysis reactions. Examples are the digestive enzymes such as sucrase, amylase, maltase, and lactase. They are classified according to the type of bond hydrolysed 1 for ester 2 for glycosidic bond, 3 for ether bond, 4 for peptide bond etc. third digit denotes the type of bond hydrolysed like 1 for hydrolysis of ester bond, 2 for thiol ester bond, 3 for monoester bond, 4 for phosphoric diester hydrolases. For example alkaline phophatases is a hydrolase that hydrolyse variety of substrate at alkaline pH. 4. Lyases- These enzymes catalyze the removal of groups and create double bond in non-aqueous media. An example would be the decarboxylases. Its second digit denotes the bond broken and third digit denotes type of group removed like 1 for carboxyl group and 2 for aldehyde group and 3 for ketoacid group. L histidine carboxy lyase histidine ------------------------------------
histamine + CO2
5. Isomerases- Enzymes that catalyze the isomerization of molecules means L isomer will be D isomer after catalysis and cis isomer will be trans isomer after the catalysis for Examples L alanine will be converted into the D alanine by alanine racemases. 2nd digit refers to the type of reaction like 1 for recemization or epimerization (if inversion occurs at assymitric carbon having four different attached group. 6. Ligases- These are also called synthetases which are enzymes that catalyze condensation reactions where smaller molecules are connected with the resulting removal of a water molecule. This is accompanied with the formation of a high energy Phosphate link that stores energy. An example would be the amino acid RNA Ligases. Subclass Name EC 1 Oxidoreductases EC 1.1 Acting on the CH-OH group of donors EC 1.2 Acting on the aldehyde or oxo group of donors EC 1.3 Acting on the CH-CH group of donors EC 1.4 Acting on the CH-NH2 group of donors EC 1.5 Acting on the CH-NH group of donors EC 1.6 Acting on NADH or NADPH EC 1.7 Acting on other nitrogenous compounds as donors EC 1.8 Acting on a sulfur group of donors EC 1.9 Acting on a heme group of donors EC 1.10 Acting on diphenols and related substances as donors
EC 1.11 Acting on a peroxide as acceptor EC 1.12 Acting on hydrogen as donor EC 1.13 Acting on single donors with incorporation of molecular oxygen (oxygenases) EC 1.14 Acting on paired donors, with incorporation or reduction of molecular oxygen EC 1.15 Acting on superoxide radicals as acceptor EC 1.16 Oxidising metal ions EC 1.17 Acting on CH or CH2 groups EC 1.18 Acting on iron-sulfur proteins as donors EC 1.19 Acting on reduced flavodoxin as donor EC 1.20 Acting on phosphorus or arsenic in donors EC 1.21 Acting on X-H and Y-H to form an X-Y bond EC 1.97 Other oxidoreductases EC 2 Transferases EC 2.1 Transferring one-carbon groups EC 2.2 Transferring aldehyde or ketonic groups EC 2.3 Acyltransferases EC 2.4 Glycosyltransferases EC 2.5 Transferring alkyl or aryl groups, other than methyl groups EC 2.6 Transferring nitrogenous groups EC 2.7 Transferring phosphorus-containing groups EC 2.8 Transferring sulfur-containing groups EC 2.9 Transferring selenium-containing groups EC 3 Hydrolases EC 3.1 Acting on ester bonds EC 3.2 Glycosylases EC 3.3 Acting on ether bonds EC 3.4 Acting on peptide bonds (peptidases) EC 3.5 Acting on carbon-nitrogen bonds, other than peptide bonds EC 3.6 Acting on acid anhydrides EC 3.7 Acting on carbon-carbon bonds EC 3.8 Acting on halide bonds EC 3.9 Acting on phosphorus-nitrogen bonds EC 3.10 Acting on sulfur-nitrogen bonds EC 3.11 Acting on carbon-phosphorus bonds EC 3.12 Acting on sulfur-sulfur bonds EC 3.13 Acting on carbon-sulfur bonds EC 4 Lyases EC 4.1 Carbon-carbon lyases EC 4.2 Carbon-oxygen lyases EC 4.3 Carbon-nitrogen lyases EC 4.4 Carbon-sulfur lyases EC 4.5 Carbon-halide lyases EC 4.6 Phosphorus-oxygen lyases EC 4.99 Other lyases EC 5 Isomerases EC 5.1 Racemases and epimerases EC 5.2 cis-trans-Isomerases EC 5.3 Intramolecular isomerases EC 5.4 Intramolecular transferases (mutases) EC 5.5 Intramolecular lyases EC 5.99 Other isomerases EC 6
Ligases EC 6.1 EC 6.2 EC 6.3 EC 6.4 EC 6.5 EC 6.6 SN 1 2 3 4 5 6
Forming Forming Forming Forming Forming Forming
carbonoxygen bonds carbonsulfur bonds carbonnitrogen bonds carboncarbon bonds phosphoric ester bonds nitrogenmetal bonds
Class Reaction catalyzed.
Oxidoreductases Transferases Hydrolases Lyases Isomerases Ligase Electron transfer Group transfer Hydrolysis Addition of group to double bonds Transfer of group to yield isomeric forms. Formation of C-C,C-S,C-O,C-N. Some metalloenzymes and their cofactor Enzyme Cofactor Cytochrome oxidase Catalase Peroxidase. Cytochrome oxidase Carbonic anhydrase Alcohol dehydrogenase Hexokinase Glucose -6-phosphate
Pyruvate kinase. Arginase Ribonucleotide reductase Pyruvate kinase. Urease Dinitrogenase Glutathione peroxidase Fe+2 or Fe+3
Cu+2 Zn+2 Mg+2
Mn+2 K+ Ni+2 Mo Se In addition to the classification name most enzymes have group names. Hydrolases-Enzymes that catalyze hydrolysis reactions. hydrogenases- Enzymes that catalyze the addition of Hydrogen atoms Oxidases- Enzymes that catalyze oxidations In addition each enzyme has a specific name which usually consists of part of the name of the substrate molecule. Examples are sucrase, Maltase, Lactase, galactosidase, and RNAase. Other enzymes have retained common names attributed to them. Enzymes such as, Pepsin, Chymotrypsin, Trypsin, Catalase, Alpha Amylase, Lysozyme, and others. Chapter-2 Types of Enzymes
A. Simple enzymes They are composed solely of protein, single or multiple subunits. If they consist of single polypeptide unit they are termed as monomeric enzyme ( sometime like in chymotrypsinogen they are of three polypeptide and are inctive zymogen form and
after cleavage from pie to delta and finally to alpha trypsinogen they become active and consist of two polypepide and still they are termed as monomeic enzyme since they are read in continous sequences). Monomeric enzymes are of few 200-300 amino acid and molecular weight ranges from 15,000 kd to 35,000 kd (kilodalton) like trypsin , elastases, that are termed as serine proteases because of an active serine residue. Serine residue become active due to relay mechanism of electron transfer (adjacent amino acid can transfer electron in series of pathway and make the serine active sufficient to active at electron deficient or carbocation center of substrate making breaking of bond possible. Thus a slight decrease in entropy make free energy negative that is essential for stabilization of intermediate transition state). Other enzyme consisting of two or more polypeptide are termed as oligomeric enzyme. Like alcohol dehydrogenase (contains two polypeptide and Zn+2 and act by electrostatic meechansim) and lactate dehydrogenase that requires cofactor NAD+ or NADP+ for its activity and occurs in multiple unit like H4, H3M, H2M2, HM3, and M4. Thus they contain four subunit and substrate can bind to each of four subunit and they are termed as isozyme. Actually Ogstien considered three sites on enzyme one or two binding site along with one catalytic subunit. Lactate is formed from pyruvate in aneorbic condition and thus pyruvate accumulation are avoided. Lactate arev then transported to other organs. B. Complex enzymes Complex enzymes They involve protein plus small organic molecule(s) or metal. Entire complex called holoenzyme. Protein part called apoenzyme. Non-protein part called coenzyme or prosthetic Group. They form the integral part of the enzyme and remain adjacent to the catalytic center to provide coordination for interacting with substrate functional group. Therefore multiple substrates can interact with enzyme and react with them. The detailed mechanism will be discussed in next topic under the head of enzyme specificities. These rules give each enzyme a unique number. Enzymes are also classified on the basis of their composition. Enzymes composed completely of protein are known as simple enzymes in contrast to complex enzymes, which are composed of protein plus a relatively small organic molecule. Complex enzymes are also known as holoenzymes. In this terminology the protein component is known as the apoenzyme, while the non-protein component is known as the coenzyme or prosthetic group where prosthetic group describes a complex in which the small organic molecule is bound to the apoenzyme by covalent bonds; when the binding between the apoenzyme and non-protein components is non-covalent, the small organic molecule is called a coenzyme. Many prosthetic groups and coenzymes are water-soluble derivatives of vitamins. It should be noted that the main clinical symptoms of dietary vitamin insufficiency generally arise from the malfunction of enzymes, which lack sufficient cofactors derived from vitamins to maintain homeostasis. The non-protein component of an enzyme may be as simple as a metal ion or as complex as a small non-protein organic molecule. Enzymes that require a metal in their composition are known as metalloenzymes if they bind and retain their metal atom(s) under all conditions that is with very high affinity. Those have a lower affinity for metal ion, but still require the metal ion for activity, are known as metal-activated enzymes. Role of Coenzymes The functional role of coenzymes is to act as transporters of chemical groups from one reactant to another. The chemical groups carried can be as simple as the hydride ion (H+ + 2e-) carried by NAD or the mole of hydrogen carried by FAD; or they can be even more complex than the amine (-NH2) carried by pyridoxal phosphate. Since coenzymes are chemically changed as a consequence of enzyme action, it is often useful to consider coenzymes to be a special class of substrates, or second substrates, which are common to many different holoenzymes. In all cases, the coenzymes donate the carried chemical grouping to an acceptor
molecule and are thus regenerated to their original form. This regeneration of coenzyme and holoenzyme fulfills the definition of an enzyme as a chemical catalyst, since (unlike the usual substrates, which are used up during the course of a reaction) coenzymes are generally regenerated. A. Catalysts Catalysts are substances that increase product formation by lowering the energy barrier (activation energy) for the product to form and increase the favorable orientation of colliding reactant molecules for product formation to be successful. There are two types of catalysts: 1. Heterogeneous Catalysts 2. Homogeneous Catalysts Heterogeneous catalysts are those that provide a surface for the reaction to proceed upon. The catalyst and the reactant molecules are not in the same phase. This is sometimes referred to as surface catalysts. Certain transition state metals like Palladium, Platinum, Nickel, and Iron serve as industrial catalysts that catalyze a wide variety of reactions such as Hydrogenation. Homogeneous catalysts are catalysts that exist in the same phase as the reactant molecules usually in a solution. Acids and Bases in solution serve as catalysts in a wide variety of Organic reactions. Most industrial catalysts are responsible for more than one catalysis among reactants and are considered relatively non-specific in what they catalyze. Enzymes are very different. All current research suggests that enzymes are extremely specific in what a given enzyme catalyzes. Indeed, most enzyme molecules catalyze only one specific reaction but it does so in a phenomenally efficient manner. One enzyme molecule might be responsible for converting thousands of reactant molecules called substrate molecules into product. B. Biocatalyst or Enzymes Enzymes are the protenaceous catalyst of the biosphere. Enzymes increase the rate of reactions without involving themselves being altered in the process of substrate conversion to product They mediate the formation of several biomolecules like Polynucleotide (DNA, RNA) protein enzyme, starch in plants, ATP etc. The Biological Catalysts are efficient in lowering down the activation energy of the reaction and thus they occur at very low temperature and pressure. All the chemical reactions inside the cell take place with the help of enzyme. Enzyme mediates rearrangement of the bond in substrate molecule by breaking of one covalent bond and formation of other covalent bond. The bond of the substrate molecule comes under strain due to presence of several functional group of different type over amino acid residue present inside the active site of the enzyme. Enzymes are biological catalysts responsible for supporting almost all of the chemical reactions that maintain animal homeostasis. Because of their role in maintaining life processes, the assay and pharmacological regulation of enzymes have become key elements in clinical diagnosis and therapeutics. The macromolecular components of almost all enzymes are composed of protein, except for a class of RNA modifying catalysts known as ribozymes. Ribozymes are molecules of ribonucleic acid that catalyze reactions on the Phosphodiester bond of other RNAs. Enzymes are found in all tissues and fluids of the body. Intracellular enzymes (enzyme located inside the cell and can be isolated after breaking the wall of the cell) catalyze the reactions of metabolic pathways. Plasma membrane enzymes (membrane bound enzyme) regulate catalysis within cells in response to extracellular signals (signal may be any chemical or any substrate molecule), and enzymes of the circulatory system are responsible for regulating the clotting of blood. Almost every significant life process is dependent on enzyme activity.
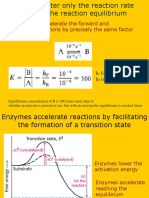
Enzyme may occur anywhere inside the cell, like cell cytoplasm, vacuoles, and cell organelle. They are found in all the living system except the viruses. Enzymes are active at certain optimum temperature, pH. Enzymes can be active at higher temperature and pH if isolated from the extremophiles bacteria living in the harsh condition. One such enzyme is utilized in the PCR polymerase chain reaction, the taq polymerase isolated from the thermus aquaticus. Enzymes mostly mediate formation of specific isomer either L (+) or D (-). They are specific towards the substrate and remains in inactive form inside the cell. Their isolation involves the breaking of cell membrane, and their supernatant is utilized for further purification and isolation by NH4 SO4. In recent years Enzymes are in more demand due to their application in various biotech and pharmacy industry. In old days renine (new name chymosine) were utilized for the formation of cheese from milk. Most of the biological molecules are stable in the neutral pH, mild temp, and aqueous environment, found inside the cell. An enzyme provides specific environments found inside the cells. They have pocket like structure called as active sites ( is only small portion of total protein and contains substrate binding site and catalytic site. Catalytic site contains various amino acid residue which reacts with substrate and form a complex structure). Molecule is bounded by the active sites and acted upon by the enzyme is called as substrate. Most of the study done regarding the ES complex since this complex decides overall rate of reactions means formation of products. Actually an enzyme decreases the free energy between the ground state and the transition state. This difference of energy is called as activation energy. Higher activation energy reflects the slower reaction. The rate of reaction can be increases by increasing the temperature. Catalyst or enzymes decreases this requirement by lowering down the activation energy. Enzymes never disturb the equilibrium. See graph in the figure. Transition state. ES Free energy G Enzyme lowers the activation energy. E P Reaction coordinate R= reactant P = product G = H T S this is the thermodynamical term used to explain the stability of reactions. A negative free energy stabilizes the system and makes the reaction possible. H denotes the enthalpy and S denotes the entropy. In unstabilized or uncatalysed system S is positive and formation of transition complex renders them lowering of entropy leading to make overall free energy negative. A more ordered system also has lower entropy for example free amino acid has more entropy than the amino acid linked with the peptide bond. How enzymes speed up the reaction without taking part? In cells and organisms most reactions are catalyzed by enzymes, which are regenerated during the course of a reaction. These biological catalysts are physiologically important because they speed up the rates of reactions many folds (up to 10 6 to 10 15 times than normal one) that would otherwise be too slow to support life. Enzymes increase reaction rates--- sometimes by as much as one millionfold, but more typically by about one thousand fold. Catalysts speed up the
forward and reverse reactions proportionately so that, although the magnitude of the rate constants of the forward and reverse reactions is are increased, the ratio of the rate constants remains the same in the presence or absence of enzyme. Since the equilibrium constant is equal to a ratio of rate constants, it is apparent that enzymes and other catalysts have no effect on the equilibrium constant of the reactions they catalyze. Enzymes increase reaction rates by decreasing the amount of energy required to form a complex of reactants that is competent to produce reaction products. This complex is known as the activated state or transition state complex for the reaction. Enzymes and other catalysts accelerate reactions by lowering the energy of the transition state. The free energy required to form an activated complex is much lower in the catalyzed reaction. The amount of energy required to achieve the transition state is lowered; consequently, at any instant a greater proportion of the molecules in the population can achieve the transition state. The result is that the reaction rate is increased. Unit of activity Micromole substrate consumed or product formed per minute is referred as IU international unit. SI unit is katal that is defined as Micromole substrate consumed or product formed per second.
Chapter 3 Enzyme Specificity Although enzymes are highly specific for the kind of reaction they catalyze, the same is not always true of substrates they attack. For example, while succinic dehydrogenase (SDH) always catalyzes an oxidation-reduction reaction and its substrate is invariably succinic acid, alcohol dehydrogenase (ADH) always catalyzes oxidation-reduction reactions but attacks a number of different alcohols, ranging from methanol to butanol. Generally, enzymes having broad substrate specificity are most active against one particular substrate. In the case of ADH, ethanol is the preferred substrate. Enzymes also are generally specific for a particular steric configuration (optical isomer) of a substrate. Enzymes that attack D sugars will not attack the corresponding L isomer. Enzymes that act on L amino acids will not employ the corresponding D optical isomer as a substrate. The enzymes known as racemases
provide a striking exception to these generalities; in fact, the role of racemases is to convert D isomers to L isomers and vice versa. Thus racemases attack both D and L forms of their substrate. As enzymes have a more or less broad range of substrate specificity, it follows that a given substrate may be acted on by a number of different enzymes, each of which uses the same substrate(s) and produces the same product(s). The individual members of a set of enzymes sharing such characteristics are known as isozymes. These are the products of genes that vary only slightly; often, various isozymes of a group are expressed in different tissues of the body. The best studied set of isozymes is the lactate dehydrogenase (LDH) system. LDH is a tetrameric enzyme composed of all possible arrangements of two different protein subunits; the subunits are known as H (for heart) and M (for skeletal muscle). These subunits combine in various combinations leading to 5 distinct isozymes. The all H isozymes is characteristic of that from heart tissue, and the all M isozymes is typically found in skeletal muscle and liver. These isozymes all catalyze the same chemical reaction, but they exhibit differing degrees of efficiency. The detection of specific LDH isozymes in the blood is highly diagnostic of tissue damage such as occurs during cardiac infarct. Thus enzyme may be group specific if they act over on several closely related substrate for example alcohol dehydrogenase on which several alcohol are substrate, or act over by specific substrate like glucokinase that transfer phosphate from ATP to glucose. They exhibit absolute specificity. They may be of product specific or substrate specific or stereospecific act on specific stereo molecules like alanine recemase acts on only Lalanine to convert them into Dalanine. Specificity is actually decided by amino acid residue present in the active site of enzyme that is capable of binding to the substrate molecule. The amino acids that are not involved in binding to the substrate do not have any contribution towards the specificity. Therefore both the subunit of enzyme like binding and catalytic site is the active center of the enzymes. They are only small part of the total enzyme and they must be accessible to the substrate. For example chymotrypisn are inactive because their zymogen form dont have accessible catalytic center. Therefore further cleavage occurs to make them active exposing the catalytic center. For proper catalytic activity their side chains must be of suitable size, shape, and character and must not block the substrate from binding. For example of several seine proteases like chymostrypsinogen, trypsin and elastases having almost similar structure but differ in specificity due to variations in the side chain amino acids. In Elastases substrate are blocked from binding due to presence of two bulky side chain valine and threonine while chymostrypsin and trypsin have no bulky group but instead contains glycine amino acid that facilitates the substrate to approach easily. Replacement of glycine by alanine make the enzyme more active. Active site most often contains both polar as well as non-polar amino acid thus facilitates the interaction with both hydrophilic and hydrophobic amino acid. This is the reason why sometime in presence of non-polar solvent like DMSO dimethylsulphoxide some enzyme are more reactive. Other examples include chymotrypsin, alcohol dehydrogenase, etc., ie. a variety of enzymes from different classes have been shown to function in nonaqueous media. Specificity May Be Very Strict Or Relatively Broad Broad specificity is shown by many degradative enzymes e.g. alkaline phosphatase, which removes phosphates from a wide range of molecules, or carboxypeptidase which snips the C-terminal amino-acid off many polypeptide chains. Intermediate specificity is most common e.g. alcohol (ethanol) dehydrogenase of E. coli will act on C2, C3, and C4 alcohols; pyruvate dehydrogenase will also use the C4 homolog of pyruvate etc.Some enzymes are absolutely specific. Aspartase catalyses the interconversion of aspartate and fumarate: L-Aspartate Fumarate + NH3
Fig . showing asymmetric binding site in the enzymes. The model of enzyme structure was proposed by fisher as three point one catalytic site and others are binding site. Sometime enzymes have both catalytic as well as binding site as same site. Such enzymes follows michaelis behaviour while others having complex and separate site behave in more complex way as allosteric site. It will only use aspartate (not similar amino acids such as glutamate) and only the L-isomer. Nor will it add NH3 to maleate, the cis isomer of fumarate.
Three theory were proposed to explain the specificity of enzyme but all of them are correct and none of them are alone operative in the enzyme specificity. 1Lock and key hypothesis 2Induced fit hypothesis. 3Transition state stabilization ENZYME MECHANISM MODELS There are two major considerations - substrate specificity and catalytic power. Enzymes must bind the correct substrate and position it correctly relative to catalytically active groups in the active site. Most enzymes are very large relative to their substrates. The actual reaction occurs in the active site, a small region of the enzyme where the substrate is bound. A. Lock and Key Model for Enzyme Activity The high specificity and efficiency of enzymes can be explained by the manner that they associate with the reactant molecules called the substrate. One of the first theories or models that help to explain this phenomenally efficient catalytic efficiency of enzymes is called the "lock and key" model. According to this model, each enzyme molecule may have as few as one active site on the surface of the enzyme molecule itself. An active site is an indentation or cavity whereby a reactant molecule (substrate) is attached to. This is called the enzyme-substrate complex. The polar and non-polar groups of the active site attract compatible groups on the substrate molecule so that the substrate molecule can effectively lock into the cavity and position itself for the necessary collisions and bond breaks and formations that must take place for successful conversion to a product molecule. Once the product molecule has been formed the electrical attractions that made the substrate molecule adhere to the active site no longer are present, and the product molecule can disengage itself from the active site thus freeing the site for another incoming substrate molecule. This process occurs in a highly efficient manner hundreds or even thousands of times in a short time span. This model assumes that molecules that lock into the active site must form a perfect fit. Also the assumption is that the active site conformation is ridged. Evidence does not support these assumptions. For example, certain molecules can lock into the active site even though the bogus molecules have a different shape compared to the true substrate molecule. This has the effect of inhibiting enzyme activity. This does not seem to support the ridged active site assumption in the lock and key model. Furthermore, small temperature changes and small changes in pH will not result in the enzyme being inhibited from catalyzing its intended reaction. The occurrence of pH and temperature ranges of optimum enzyme activity does not support the assumptions made by the lock and key model of ridged active site cavities. Modification of the lock and key model is necessary to account for the occurrence of pH and temperature ranges of optimum enzyme activity and to explain why other molecules can effectively block the active site. B. Induced Fit (Hand and Glove) Model of Enzyme Activity Modification of the lock and key model assumes that the active site has a certain amount of elasticity whereby the active site can expand or contract in a limited way in order to accommodate the substrate molecule. The analogy is like a hand
fitting into a glove. The glove adjusts in shape and size to fit various sized hands within a certain range. This tolerance would explain why bogus molecules of slightly different size compared to the true substrate molecule can still be accommodated by the elastic active site. Small changes in temperature would distort the active site conformation but not so much that the active site could not still accommodate the substrate molecular size. PH changes which would also change the active site conformation but not so much that the active site could not flexibly accommodate the substrate molecule. The Induced Fit Model seems to explain why there is some flexibility in the abilility of the active site to accommodate other molecules and at limited temperature and pH ranges. Induced fit assumes that the active site of an enzyme is not complementary to that of the transition state in the absence of the substrate. Such enzymes will have a lower value of kcat/KM (a numerical value to compare the catalytic efficiency of enzyme), because some of the binding energy must be used to support the conformational change in the enzyme. Induced fit increases KM without increasing kcat. Induced fit example Hexokinase catalyzes the ATP-dependent phosphorylation of glucose. The binding of glucose to the enzyme induces a major conformational change - the induced fit. Phosphoglycerate kinase catalyzes the synthesis of ATP. The two substrates are brought together in the active site by a conformational change. An induced fit at an interface. Lipases are enzymes that hydrolyze esters of long chain fatty acids. These enzymes show virtually no activity against water-soluble substrates and only hydrolyze substrates present in the interface between water and lipid interfacial activation. The activation of the lipases at the interface is a result of a conformational change, induced fit, caused by the enzyme binding to the interface. This conformational change exposes the active site of the enzyme. Mechanisms of Enzyme Catalysis Enzymes are proteins. Every protein is determined by its amino acid sequence (its primary structure) and its tertiary structure (the three-dimensional folding of the polypeptide chain). Its uniqueness is caused by the sequence and nature of its amino acid side chains. They form a number of weak interactions, which again are the basis for the spatial arrangement (conformation) of the molecule and which help to maintain this structure by stabilizing it. In that way, the onedimensional information that was stored in the genome as a DNA sequence is first transcribed into mRNA and after translation into an amino acid sequence finally transformed into a three-dimensional structure. It is this structure that allows an enzyme to perform its catalytic activity. That explains the specificity of catalysis and the selectivity for a certain substrate (and also that for additional regulatory factors). Enzyme molecules are, compared to most of their substrate molecules, rather large. Their surface is not evenly structured but displays dent-ins, grooves, pockets, hollows etc. The part that binds to a substrate molecule is termed the active centre or substrate-binding site and is characterized by a shape complementary to that of the substrate molecule. Furthermore, certain amino acid side chains are exposed at the active centre. They are engaged in the catalytic turn-over of the substrate. Numerous enzymes depend on additional factors that are necessary to perform the catalytic reaction. In other words: the enzyme binds first to a coenzyme like NAD or FAD. Often, the enzyme's surface is structured in a way that the coenzyme is bound to a specifically shaped pocket. Depending on the type of molecule, the binding is either reversible (achieved through weak interactions) or irreversible (covalent bonds). The shape of the holoenzyme (= apoenzyme [protein] + coenzyme) causes the substrate selectivity. Atoms or ionized groups of the coenzyme take part in the catalytic reaction. These properties explain, why reactions take place at an enzyme's surface that occur in solutions only after a considerable amount of activation energy has been
supplied. The binding sets reactants into a state of enhanced reactivity recognizable by the close vicinity and the right orientation towards each other. The collision theory states that the event of two molecules meeting in solution in a way that a bond can form between them is rather improbable, though the probability can be considerably increased by the supply of energy (pressure, temperature).
Chapter 4 Enzyme-Substrate Interactions The favored model of enzyme substrate interaction is known as the induced fit model. This model proposes that the initial interaction between enzyme and substrate is relatively weak, but that these weak interactions rapidly induce conformational changes in the enzyme that strengthen binding and bring catalytic sites close to substrate bonds to be altered. After binding takes place, one or more mechanisms of catalysis generate transition- state complexes and reaction products. The possible mechanisms of catalysis are five in number: 1. 2. 3. 4. 5. Catalysis by approximation General acid, general base catalysis Catalysis by electrostatic effects Covalent catalyis (nucleophilic or electrophilic) Catalysis by strain or distortion
a. Catalysis by Bond Strain In this form of catalysis, the induced structural rearrangements that take place with the binding of substrate and enzyme ultimately produce strained substrate bonds, which more easily attain the transition state. The new conformation often forces substrate atoms and bulky catalytic groups, such as aspartate and glutamate, into conformations that strain, existing substrate bonds. b. Catalysis by Proximity and Orientation Enzyme-substrate interactions orient reactive groups and bring them into proximity with one another. In addition to inducing strain, groups such as aspartate are frequently chemically reactive as well, and their proximity and orientation toward the substrate thus favors their participation in catalysis. The classic way that an enzyme increases the rate of a bimolecular reaction is to use binding energy to simply bring the two reactants in close proximity. If DG is the change in free energy between the ground state and the transition state, then DG=DHtDS. In solution, the transition state would be significantly more ordered than the ground state, and DS would therefore be negative. The formation of a transition state is accompanied by losses in translational entropy as well as rotational entropy. Enzymatic reactions take place within the confines of the enzyme active-site wherein the substrate and catalytic groups on the enzyme act as one molecule. Therefore, there is no loss in translational or rotational energy in going to the transition state. This is paid for by binding energy.
c. Catalysis Involving Proton Donors (Acids) and Acceptors (Bases) Other mechanisms also contribute significantly to the completion of catalytic events initiated by a strain mechanism, for example, the use of glutamate as a general acid catalyst (proton donor). General acid-base catalysis is involved in a majority of enzymatic reactions. General acidbase catalysis needs to be distinguished from specific acidbase catalysis. Specific acidbase catalysis means specifically, OH or H+ accelerates the reaction. The reaction rate is dependent on pH only, and not on buffer concentration. In General acidbase catalysis, the buffer aids in stabilizing the transition state via donation or removal of a proton. Therefore, the rate of the reaction is dependent on the buffer concentration, as well as the appropriate protonation state. In the second step (collapse of the tetrahedral intermediate), the leaving group must be protonated. The general acidbase is best when its pKa is near that of the pH of the solution, in order to have appropriate concentrations of each buffer species. Specific acid-base catalysis is due to H+ or OH- ions which temporarly donates proton or accept proton. General acid-base catalysis is due to proton donors or proton acceptors, which donate or remove protons to (or from) the transition state intermediate. Most hydrolytic enzymes use acid/base catalysis. General acid-base catalysis is a commonly employed mechanism in enzyme reactions, e.g., hydrolysis of ester/ peptide bonds, phosphate group reactions, addition to carbonyl groups, etc. General acid catalysis involves donation of a proton by the catalyst. General base catalysis involves abstraction of a proton by the catalyst. The side chains of Asp, Glu, His, Cys, Tyr and Lys can be involved in general acid-base catalysis. Concerted acid-base catalysis Providing both acid and base simultaneously is impossible in free solution, yet enzymes can do this. An acidic group in one part of the active site donates a proton and a basic group in another part of the active site removes a second proton from the reaction intermediate. Amino acids with extra carboxyl or amino groups can obviously act as acids or bases. In addition, histidine (pK around 7.0) can act as either an acidic or basic catalyst depending on whether or not it is protonated. Also, histidine reacts very fast (protonation/deprotonation of imidiazole ring has a reaction half-time of less than 10-10 seconds at neutral pH). Histidine is rarely found in proteins except at catalytic sites. d. Covalent Catalysis: In catalysis that takes place by covalent mechanisms, the substrate is oriented to active sites on the enzymes in such a way that a covalent intermediate forms between the enzyme or coenzyme and the substrate. One of the best-known examples of this mechanism is that involving proteolysis by serine proteases, which include both digestive enzymes (trypsin, chymotrypsin, and elastase) and several enzymes of the blood clotting cascade. These proteases contain an active site serine whose R group hydroxyl forms a covalent bond with a carbonyl carbon of a peptide bond, thereby causing hydrolysis of the peptide bond. Biologically important nucleophiles are negatively charged or contain unshared electrons Biologically important electrophiles generally are either positively charged, or contain unfilled valence electron shells, or contain an electronegative atom
Covalent Intermediates Here the strategy is to lower the transition state energy "hump" by taking an alternative reaction pathway therefore sometime it is also called as alternative pathway catalysis. Sometime nucleophilic catalyst form intermediate more rapidly than normal catalysis. In covalent catalysis, a covalent bond is transiently formed between the substrate and the enzyme (or coenzyme). This reaction usually involves a nucleophilic group on the enzyme and an electrophilic group on the substrate Uncatalysed: BX + Y BY + X Catalysed: BX + Enz Enz-B + X Enz-B + Y Enz + BY Where, B is usually some chemical group such as a phosphate group, acyl group or glycosyl group. Phosphoenzymes usually attach the phosphate to serine or histidine. Serine type - phosphoglucomutase, alkaline phosphatase. Histidine type - glucose 6 phosphatase, phosphotransferase system. Acyl enzymes usually attach the acyl group at an active serine or cysteine residue. Most proteases (eg trypsin, elastase, subtilisin are serine enzymes. Cysteine enzymes include papain (a protease), glyceraldehyde phosphate dehydrogenase and most enzymes using acyl CoA derivatives. The proteases are globular, water soluble proteins that function as enzymes. They catalyze the hydrolyses of peptide bonds in proteins. Being enzymes, proteases can be characterized by their substrate affinity and the catalytic rate of the reaction. Using proteases to study the effects of single amino acid substitutions (mutations) on catalytic rate and substrate affinity demonstrated that these two properties are linked and that this linkage can be explained by analyzing the conformation of the catalytic or active site of the enzyme. This analysis showed that four major functional groups are found in the catalytic site of proteases and based on this functional groups, 4 families of proteases have been defined: a. Serine proteases b. Cystein proteases c. Aspartate proteases d. Metallo proteases This classification uses the functional group within the enzyme, and does not relate to the substrate specificity of the proteases themselves. The name of the protease family refers to an amino acid (e.g. aspartate) or a metal as co-enzyme at the active site of the enzyme. Structural element of active site Function The main chain substrate binding unspecific binding of polypeptide segment Specificity pocket semi-specific binding of side chains, sequence specificity Oxyanion whole stabilizes S* over S in enzyme The catalytic triad forms tetrahedral intermediate (transition state; stabilizes S* over S); hydrolyzes peptide bond The serine proteases are a class of enzymes that degrade proteins in which a serine in the active site plays an important role in catalysis. The family includes among many others, Chymotrypsin and trypsin, which weve talked about, and Elastase. All three enzymes are similar in structure, and they all have three important conserved residuesa histidine, an aspartate, and a serine. Thrombin is crucial in the blood-clotting cascade Subtilisin is bacterial protease Plasmin breaks down the fibrin polymers of blood clots Tissue plasminogen activator (or TPA), cleaves the
proenzyme plasminogen, yielding plasmin
The six step reaction mechanism of chymotrypsin: 1. Substrate attaches to enzyme at the main chain binding site and specificity pocket with the scissile (peptide) bond exposed to the triad (ES formation) 2. covalent bond formation between -C=O and S195 hydroxyl (nucleophilic attack), tetrahedral intermediate as transition state, oxyanion hole stabilization, H57 binds the -released H+ from S195 (ES*) 3. acyl-enzyme intermediate and peptide bond hydrolysis, peptide with new N-term released from enzyme taking -H from H57 (originally S195; see step 2) (EP*) 4. A water molecule placed next to H57 and acyl-enzyme intermediate at S195 (EP*) 5. the water molecule initiates a nucleophilic attack and undergoes a reaction with one -H attaching to H57 and -OH to acyl-enzyme intermediate forming again a tetrahedral transition state of the new C-terminal peptide fragment (EP*) 6. Peptide released while H57 donates -H (from water molecule) to S195 (E+P) Chymotrypsin cleaves after mainly aromatic amino acids, while trypsin cleaves after basic amino acids. Elastase is fairly nonspecific, and cleaves after small neutral amino acids. Notice how their active sites are suited for these tasks. Chymotrypsin is a protease that cleaves peptide bonds on the carboxyl side of aromatic or large hydrophobic amino acids the mechanism of chymotrypsins cleavage reaction includes covalent, general acid-base, electrostatic, and proximity/orientation catalysis The tetrahedral intermediate in chymotrypsin, which consists of the Ser195 adduct before departure of the leaving group, is considered to be the transition state intermediate in the chymotrypsin reaction. It is high energy because there is a carbon surrounded by 3 electronegative atoms, one of which bears a negative charge. How is it that the enzyme stabilizes this transition state intermediate? The backbone amides of gly193 and ser195 form an oxyanion hole. They loosely hydrogen bond to the carbonyl oxygen under attack. Upon formation of the tetrahedral intermediate, the resulting carbon-oxygen single bond is longer, and the negatively charged oxygen is better accommodated in the oxyanion hole. Diisopropylflurophosphate is an inhibitor of chymotrypsin. It diffuses into the active, wherein a nucleophilic amino acid attacks the phosphate, releasing fluoride anion. This results in a covalent bond between the nucleophile and the inhibitor. It inhibits the reaction because it blocks entry of normal substrates. The enzyme-inhibitor adduct is very stable. Upon hydrolysis of the protein (6 N HCl, 110C) and amino acid analysis on the hydrolysate, a novel amino acid was isolated. It was the diisopropylphosphoryl derivative of serine. Crystal structures of chymotrypsin reveal the reason for the high reactivity of Ser 195 Ser 195 is in a H-bonding interaction with His 57, which is H-bonding with Asp 102 (catalytic triad) the catalytic triad serves to activate Ser 195 through general acid-base catalysis catalytic triads are common in serine proteases Conformation changes from trigonal to tetrahedral in the peptide upon nucleophilic addition allow the oxyanion formed to move into a hole with two backbone amides inside the oxyanion hole the transition state anion can form two hydrogen bonds with the amide hydrogens as donors the enzyme cannot form H-bonds with the amide hydrogens in the oxyanion hole when the peptide is in trigonal conformation The mechanism of chymotrypsin utilizes stabilization of the transition state, covalent catalysis, acid-base catalysis, electrostatic, proximity and orientation effects Substrate specificities in proteases are due to active site binding pockets. Chymotrypsin substrate specificity is due almost entirely to one deep, hydrophobic pocket that binds the sidechain of the amino acid N-terminal to the site of bond cleavage (S1) binding of appropriate sidechains to this pocket positions the adjacent peptide bond into the active site for cleavage Many serine
proteases have high similarity to chymotrypsin trypsin and elastase are approximately 40% identical in primary sequence to chymotrypsin and their overall structures are very close Substrate specificity of these other proteases compared to chymotrypsin can be attributed to their S1 binding pockets Trypsin: cleaves after positively charged residues, S1 has Asp Elastase: cleaves after small side chains (A, S), S1 has bulky Val residues
Histidine, serine and cysteine operate by nucleophilic catalysis. Their nucleophilic groups (imidazole ring, OH and SH respectively) are good electron donors the intermediates they form are unstable and react easily with the final acceptors. Since the whole point is that covalent intermediates should rapidly break down to release the reaction product, enzyme covalent intermediates are very difficult to isolate. cystein prtoease Renin - An Aspartyl Protease A Metalloprotease with Substrate Carbonic Anhydrase Mechanism His64 assists in H+ removal HCO3- released. H2O enters HO- attacks CO2
Methylation prevents H-bonding with DNA substrate Lysozyme is a small globular protein composed of 129 amino acids. It is also an enzyme which hydrolyzes polysaccharide chains, particularly those found in the peptidoglycan cell wall of bacteria. In particular, it hydrolyzes the glycosidic bond between C-1 of N-acetyl muramic acid and C-4 of Nacetyl glucosamine. It is found in many body fluids, such as tears, and is one of the bodys defenses against bacteria. The best studied lysozymes are from hen egg whites and bacteriophage T4. Although crystal structures of other proteins had been determined previously, lysozyme was the first enzyme to have its structure determined. The X-ray crystal structure of lysozyme has been determined in the presence of a nonhydrolyzable substrate analog. This analog binds tightly in the enzyme active site to form the ES complex, but ES cannot be efficiently converted to EP. It would not be possible to determine the X-ray structure in the presence of the true substrate, because it would be cleaved during crystal growth and structure determination. The active site consists of a crevice or depression that runs across the surface of the enzyme. Look at the many hydrogen bonding contacts between the substrate and enzyme active site that enables the ES complex to form. There are 6 subsites within the crevice, each of which is where hydrogen bonding contacts with
the sugars are made. In site D, the conformation of the sugar is distorted in order to make the necessary hydrogen bonding contacts. This distortion raises the energy of the ground state, bringing the substrate closer to the transition state for hydrolysis. At what position does water attack the sugar? When the lysozyme reaction is run in the presence of H218O, 18O ends up at the C-1 hydroxyl group at site D. This suggests that water adds at that carbon in the mechanism. From the X-ray structure, it is known that the C-1 carbon is located between two carboxylate residues of the protein (Glu-35 and Asp-52). Asp-52 exists in its ionized form, while Glu-35 is protonated. Glu can act as a general acid to protonate the leaving group in the transition state. Asp can function to stabilize the positively charged intermediate. Glu then acts as a general base to deprotonate water in the transition state. Within the class of hydrolases, Lysozyme belongs to the Glycosylases family (EC 3.2.-.-). Lysozyme reaction is the hydrolysis of the beta (1-4) glycosidic bond between N-acetylglucosamine sugar (NAG) and N-acetylmuramic acid sugar (NAM) and therefore it is possible classify it as Glycosidases, i.e. enzymes hydrolyzing O- and S-glycosyl (EC 3.2.1.-) with number 17 (EC 3.2.1.17) in this group
Lysozyme Lysozyme catalyzes the hydrolysis of the (1-->4) linkage between N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) in bacterial cell wall polysaccharides and (1 4)-linked poly NAG (chitin). The lysozyme mechanism illustrates: Transition state stabilization Acid-base catalysis Several residues in the protein participate in substrate binding. The binding of NAM4 in the chair conformation is unfavorable. But the binding of residue 4 in the half-chair conformation is favorable (preferential transition state binding). Hydrolysis involves acid-base catalysis (Glu35 serves as a proton donor to the oxygen of the leaving alcohol. The resulting carbonium ion (+) is stabilized by the ionized side chain of Asp52 until it can react with water) Many glycosidases utilize a covalent intermediate in their mechanism. Here
is a good example of transition state stabilization using a bacterial glycosidase. Another excellent example that clearly shows how transition state stabilization works is found in the structure of this transferase. Chloroketones are inhibitors of serine proteases, which bind in the active site and react with His 57. Pancreas contains a small protein (6 kDa) that is a potent inhibitor of trypsin. The inhibitor is a naturally occurring transition state analog of a protein substrate. e) Metal Ion Catalysis Nearly 1/3 of all known enzymes require the presence of metals for catalytic activity Metalloenzymes - contain tightly bound metal cofactors such as Fe2+, Fe3+, Cu2+, Zn2+, Mn2+, Co2+ Metal Activated enzymes only loosely bind the metal ions. The ions are usually Na+, K+, Mg2+, or Ca2+ Many enzymes have metal ions at the active site. Sometimes these are used as redox centers, as in cytochromes, nitrate reductase etc. However, often the metal ion does not get reduced or oxidized, but acts to stabilize negative charges on the reaction intermediate. In carboxypeptidase, Zn2+ polarizes the C=O of the peptide bond which is about to be broken. In alcohol dehydrogenase Zn2+ polarizes the C=O group of acetaldehyde, so allowing the hydride ion (H) to be added to the carbon atom. Electrostatic interactions are much stronger in organic solvents than in water due to the dielectric constant of the medium. The interior of enzymes have dielectric constants that are similar to hexane or chloroform. Metal ions that are bound to the protein (prosthetic groups or cofactors) can also aid in catalysis. In this case, Zinc is acting as a Lewis acid. It coordinates to the non-bonding electrons of the carbonyl, inducing charge separation, and making the carbon more electrophilic, or more susceptible to nucleophilic attack. Metal ions can also function to make potential nucleophiles (such as water) more nucleophilic. For example, the pka of water drops from 15.7 to 6-7 when it is coordinated to Zinc or Cobalt. The hydroxide ion is 4 orders of magnitude more nucleophilic than is water. SUMMARY Metal Mn+2. Ca+2. ion catalysis is widely used. Binding substrates in the proper orientation. Mediating oxidation-reduction reactions. Electrostatically stabilizing or shielding negative charges. Metalloenzymes contain tightly bound metal ions: Fe+2, Fe+3, Cu+2, Zn+2, Metal-activated enzymes contain loosely bound metal ions: Na+, K+, Mg+2,
Electrostatic catalysis refers to the fact that when a substrate binds to an enzyme, water isusually excluded from the active site. This causes the local dielectric constant to be lower, Which enhances charge-charge interactions in the active site. Proximity and orientation effects are important in enzymatic reactions. The three dimensional structure of the enzyme can bring several reactive side chains into close proximity in the active site.
Binding of the substrate in the active site can orient the substrate for most efficient interaction with these side chains. Preferential Transition State Binding is probably the most important rate enhancing mechanism available to enzymes. This means that the enzyme binds the transition state of the reaction more tightly than either the substrate or product, therefore Go is lowered. Weak interactions between the enzyme and substrate are optimized in the transition state. Utilization of enzyme-substrate binding energy in catalysis The maximum binding energy between an enzyme and a substrate will occur when there is maximal complementarity between the structure of the binding site and the structure of the substrate. The structure of the substrate changes during the reaction, becoming first the transition state and then products. The structure of the enzyme can only be complementary to one form of the substrate. If the structure of the active site is complementary to the transition state, then binding increases as the reaction proceeds, which lowers the activation energy. Having the active site bind the transition most strongly also means that products are bound weakly, which favors dissociation of the products from the enzyme once the reaction is complete. When the active site is complementary to the transition state the full substrate binding energy is only realized as the reaction reaches the transition state, which lowers the activation energy of the reaction by the magnitude of the binding energy. Molecular Mechanisms for the Utilization of Binding Energy Strain is a classic concept in which it was supposed that binding of the substrate to the enzyme somehow caused the substrate to become distorted toward the transition state. Transition state stabilization is a more modern concept, which states that it is not the substrate that is distorted but rather that the transition state makes better contacts with the enzyme than the substrate does, so the full binding energy is not achieved until the transition state is reached. Strain or stress? It is unlikely that there is enough energy available in substrate binding to actually distort the substrate toward the transition state. It is possible that the substrate and enzyme interact unfavorably and this unfavorable interaction is relived in the transition state. We might think that the substrate is under stress meaning that it is subjected to forces but not distorted by them. It is more likely that the enzyme is strained, as for example in induced fit.
Catalytic Power Enzymes lower the energy of the transition state by stabilizing the original reaction intermediate or by providing an alternative reaction pathway. Rate increases by enzymes range from 108 to 1020 relative to the uncatalysed,
spontaneous reaction (e.g. 109 for alcohol dehydrogenase; 1016 for alkaline phosphatase). Factors involved in enzyme rate increases: a) Proximity - up to 106 fold; b)Orientation - up to 100 fold c) Covalent enzyme-substrate intermediates around 1010 fold d) General acid-base catalysis - around 1010 fold ;e) Metal ion catalysis - around 1010 fold; f) Distortion of the substrate - up to 108 fold (but largely hypothetical) a) Proximity The enzyme binds the substrate so that the susceptible bond is very close to the catalytic group in the active site or in close proximity in the active site. It increases their concentration and and reduces entropy loss for subsequent formation of a transition state. This has been called as proximation effect or approximation effect. Therefore it appears that enzyme bound transition state is the single most in determining whether the reaction should proceed. Calculations indicate that the local concentration of substrate in the active site may be as much as 50M whereas its concentration in the cytoplasm may be less than 1mM. Since chemical reaction rates are proportional to the concentrations of the reactants a rate enhancement of up to 106 may be generated by local concentration effects. This effect is counteracted to some extent by the fact that the concentration of enzyme active sites is low (the active site is a small portion of a very large molecule; the total protein concentration in cytoplasm is 1 to 10mM at most). b) Orientation Proximity alone is insufficient. The reacting groups must be properly oriented. Orbital steering hypothesis - binding of the substrate(s) to the enzyme aligns the reactive groups so that the relevant molecular orbitals overlap. This increases the probability of forming the transition state. For a typical bimolecular reaction in free solution about 1/100 collisions between molecules of sufficient energy actually leads to a reaction. Thus the maximum effect of orientation would be 100-fold. Consider the attack on an aryl ester by a carboxylic acid. Now let's put both reacting groups on the same molecule - i.e. we will link R1 and R2 together. As the five examples show, the relative rate gets greater as the reacting groups are brought closer together. In addition, holding them in the correct orientation also helps. (Ar = methoxyphenyl group.)
f. Catalysis in organic solvents Another approach that has generated some interest in enzyme technology is catalysis in non-aqueous or nearly anhydrous media (Bell et al. 1995; Carrea et al. 1995; Gupta, 1992). Alexander Klibanov & his colleagues at MIT initiated much of the work in this area. Traditionally, enzyme reactions are carried out in aqueous media. The question arises as to how much water is really needed by the enzyme to function. This led them to test how enzymes function in various amount of organic solvents. They found startling changes to enzyme properties as the water content of the reaction mixture decreases. Some of these are illustrated in overheads for porcine pancreatic lipase (Zaks & Klibanov 1984; 1985). They concluded that it is the water bound to the enzyme that is critical for activity, rather than the total water content of the system. Research by Klibanov, his colleagues and other researchers have opened up novel opportunities in enzyme technology and this field has changed from a mere curiosity to a serious alternative to chemical reactions. The functions of a number of enzymes under non-aqueous conditions have been studied. Such systems are applicable to enzymes of varying reaction types. Some examples include: 1. Lipases, which show increased thermostability, altered substrate specificity and a propensity towards synthetic reactions in non-aqueous media.
2. Horse radish peroxidase (HRP) which is able to depolymerize lignin in 95% dioxane (Dordick et al., 1986). 3. Glucose isomerase for conversion of glucose to fructose to produce high fructose corn syrup (HFCS). 4. Other examples include chymotrypsin, alcohol dehydrogenase, etc., ie. a variety of enzymes from different classes have been shown to function in nonaqueous media. Some advantages to using enzymes in non-aqueous solvents include (Dordick, 1989): 1. Increased solubility of nonpolar substrates. 2. Shifting thermodynamic equilibria to favor synthesis over hydrolysis. 3. Reduction in water-dependent side reactions. 4. Immobilization is unnecessary, because enzymes are insoluble in organic solvents, and can be recovered by simple filtration. 5. Ease of product recovery from low boiling organic solvents. 6. Enhanced thermo stability of enzymes; water is required to inactivate enzymes at high temperature. Therefore, in the absence of water, enzymes are more stable. 7. Altered activity and affinity (Km) of the enzyme. 8. Novel enzyme reactions possible (eg. new catalytic activity seen with lipases or horse radish peroxidase). 9. Elimination of microbial contamination. 10. Can use enzymes directly within a chemical process. Importance and relevance of whole cell enzymes in biotechnology Enzymes are biological catalysts. With the exception of ribozymes, enzymes are mainly proteins in nature, and may also possess lipid or sugar moieties. Their main task is to decrease the activation energy (Eact) of a chemical reaction. The whole biotechnology field depends on the activities of enzymes, either acting alone or in concert. Exploitation of enzymes is not a recent development. They have been used throughout the ages in leather tanning, in cheese-making, in the preparation of malted barley for beer brewing & the leavening of bread. These processes use enzymes in the form of whole cells. Conceptually, it is easy to decide whether one should use whole cells or isolated enzymes in a biotechnological process to produce or transform compounds. Some processes require multi-step transformations, eg. the synthesis of antibiotics, interferons, monoclonal antibodies, or ethanol production from glucose. These transformations involve a number of enzymes acting sequentially, along with the requirement for co-factor regeneration in several steps. They are clearly candidates for using whole cells. There may be some drawbacks to using whole cells in catalysis. These include 1. Competing side reactions from other enzymes. 2. Sterility problems often arise when one deals with whole cells. 3. Some conditions may lead to cell lysis, and this can result in loss of biocatalysts or severely decrease reaction rate depending on the nature and type of enzyme reaction. 4. Formation of by-products. 5. On the other hand, with one-step or two-step stereospecific transformations, enzymes can be superior because their use will eliminate some of the potential problems with using whole cells. The advantages of using enzymes include: 6. High catalytic rate possible. 7. Functions in moderate conditions. 8. Non-polluting catalysis. 9. Less by-products formed. 10. Less problem with sterility. 11. Optimizing 1 or 2 enzyme catalyzed reactions is easier than optimizing the whole pathway in a whole cell. 12. Enzymes catalyze a wide range of reactions, some with high stereospecificity, such as oxidation, reduction, dehydrogenation, dehalogenation,
hydration, dehydration, etc. Therefore, the scope of enzyme technology is very broad. 13. Despite all these striking characteristics, enzyme have not been used widely as compared to chemical catalysis. This is because the use of enzymes also suffers from some serious drawbacks: 14. Most isolated enzymes are not stable enough for use under industrial conditions. This may include poor stability with respect to temperature, pH, the presence of metal ions, etc. This is not surprising considering that most enzymes have evolved to function in a biological milieu where the conditions may differ from those used in industry. 15. In cells, all enzymes are subject to turnover, ie. old ones are replaced by new ones. This is difficult to do with isolated enzymes in vitro. 16. Most enzymes are water-soluble, and therefore, may be difficult to separate from reactants or products. 17. Many useful enzymes are intracellular, and hence are difficult to isolate. Abzymes Catalytic antibodies are also known as abzymes, are capable of catalyzing specific chemical reactions. They are able to perform this task because they have been elicited against antigen known as transition state analogues, these are molecules that closely resemble in shape and charge the reaction transition state (the highest energy spices in reaction pathway known as activated complex). Enzyme observed to bind transition complex but not the substrate, and the product. Enzyme by forming the active site similar to the transition state, enzyme stabilizes or lowers down the energy of this species. The effect is to accelerate the reaction because the activation barrier is more easily overcome. This complementarity in shape and charge to the transition state is readily demonstrated by the effectiveness of transition state analogue is a phosphonate containing molecule that mimics the transition state of the hydrolysis of the corresponding acyl derivative. When an ester is hydrolysed the central carbonyl group changes from planer sp2 structure to tetrahedral sp3. the phosphonate containing molecule analogue is able to reproduce the shape of the transition state as well as the partially negative charged oxygen. In addition, it is chemically stable, where as the actual transition state exists only fleetingly. In this example, an enzyme that catalysed the hydrolysis of this ester would also bind tightly to phosphonate analogue. The general strategies behind the production of catalytic antibody is to (i) design and synthesize a molecule whose shape is closely resemble to that of transition state of the reaction one wishes to catalyze, (2) tether this molecule to larger molecule, (3) elicit an immune response to this conjugate, (4) screen the resultant monoclonal antibodies for catalytic activity of the type desired. Antibodies that are elicited against transition state analogue and therefore have the ability to bind them will be chemically and sterically complementary to the transition state and will therefore be potentially capable of catalyzing the reaction.
Table 7. 1 Exercises LONG TYPE 1. Describe the six classes of enzyme with examples? 2. Define Biocatalyst ? Describe how biocatalyst lower down the activation energy? 3. Define Enzyme ? Classify the Enzyme on the basis of enzyme commission? 4. Define holoenzyme ? write the role of apoenzyme, cofactor, and metalloenzymes on enzyme activity. 5. Define enzyme specific activity? Write the different method of enzyme
substrate interaction in brief? Short type 1. write short notes on following a. Enzyme activity b. Enzyme specificity c. Enzyme commission. d. Holoenzyme. e. Cofactor f. Metalloenzymes. g. Coenzyme.
Module II Specificity of enzyme action, monomeric and oligomeric enzymes, Enzyme inhibition.
Chapter-5 ENZYME KINETICS & INHIBITION TERMS Enzymes are protein catalysts of biological origin that, like all catalysts, speed up the rate of a chemical reaction without being used up in the process. They achieve their effect by temporarily binding to the substrate. Competitive inhibition, when the substrate and inhibitor compete for binding to the same active site. Noncompetitive inhibition, when the inhibitor binds somewhere else on the enzyme molecule reducing its efficiency. Enzyme kinetics The study of the rate at which an enzyme works is called enzyme kinetics. Alloenzymes. If a gene exists in a heterozygous state, two different polypeptide chains will be generated. They may be separated by gel electrophoresis under favourable conditions. They may also differ in their rate of substrate turnover. The gene product a, for example, may be inactive while that of A is fully active. But all states in between are also possible. Furthermore, it was shown in numerous examples that A may be favourable under certain environmental conditions while a,
is favourable under other conditions (more in the section about evolution). Polypeptides (enzymes) that are encoded by different alleles are called alloenzymes. Isoenzymes. By duplication of the genetic material a gene may be present within a haploid genome for two or more times. It is not important whether the gene loci are on one chromosome or distributed over several chromosomes. The gene products (enzymes) of the different gene loci (pseudoalleles) are called isoenzymes. Competitive inhibitors are molecules that bind to the same site as the substrate preventing the substrate from binding as they do so - but are not changed by the enzyme. Noncompetitive inhibitors are molecules that bind to some other site on the enzyme reducing its catalytic power. INTRODUCTION Kinetics is the study of the rate of change of reactants to products .Velocity refers to the change in concentration of substrate or product per unit time. Rate refers to the change in total quantity per unit time. Initial velocity is the change in reactant or product concentration during the linear phase of a reaction. Study of rate of the catalyzed reaction is called as kinetics. This is the important step in understanding the mechanism of catalysis. Therefore, it is necessary to study the factors affecting the rate of catalysis. The rate of reactions depends on the order and molceulcarity. When enzyme concentration is small and substrate concentration increases then first the reaction occurs rapidly but maximum rate is achieved only when all the space in the active site is filled and no more substrate can be filled. This time reaction rate become slower and the product formation starts. Therefore initially the order is first and later on it becomes zero order. Therefore maximum velocity is affected by the occupying all space of the active site by the substrate. If this is occupied by the inhibitor as in competitive inhibition the v max remain same since space is same and inhibitor has similar structure as the substrate. Initially substrate consumes and product is released. This assumption is true for single substrate and single enzyme. Rate in the itegarl form is shown by the rate = dX/ dt. Chemical Reactions and Rates According to the conventions of biochemistry, the rate of a chemical reaction is described by the number of molecules of reactant(s) that are converted into product(s) in a specified time period. Reaction rate is always dependent on the concentration of the chemicals involved in the process and on rate constants that are characteristic of the reaction. For example, the reaction in which A is converted to B is written as follows: A B----------------------------1
The rate of this reaction is expressed algebraically as either a decrease in the concentration of reactant A: -[A] = k[B]-----------------------2 or an increase in the concentration of product B:
[B] = k[A]--------------------3 In the second equation (of the 3 above) the negative sign signifies a decrease in concentration of A as the reaction progresses, brackets define concentration in molarity and the k is known as a rate constant. Rate constants are simply proportionality constants that provide a quantitative connection between chemical concentrations and reaction rates. Each chemical reaction has characteristic
values for its rate constants; these in turn directly relate to the equilibrium constant for that reaction. Thus, reaction can be rewritten as an equilibrium expression in order to show the relationship between reaction rates, rate constants and the equilibrium constant for this simple case. The rate constant for the forward reaction is defined as k+1 and the reverse as k-1. At equilibrium the rate (v) of the forward reaction (A B) is by definition is equal to that of the reverse or back reaction (B A), a relationship which is algebraically symbolized as: V forward = v reverse---------------4 Where, for the forward reaction: V forward = k+1[A]---------------5 and for the reverse reaction: V reverse = k-1[B]----------------6 In the above equations, k+1 and k-1 represent rate constants for the forward and reverse reactions, respectively. The negative subscript refers only to a reverse reaction, not to an actual negative value for the constant. To put the relationships of the two equations into words, we state that the rate of the forward reaction [v forward] is equal to the product of the forward rate constant k+1 and the molar concentration of A. The rate of the reverse reaction is equal to the product of the reverse rate constant k-1 and the molar concentration of B. At equilibrium, the rate of the forward reaction is equal to the rate of the reverse reaction leading to the equilibrium constant of the reaction and is expressed by: [B]/[A] = k+1/k-1 = K eq.---------------7 This equation demonstrates that the equilibrium constant for a chemical reaction is not only equal to the equilibrium ratio of product and reactant concentrations, but is also equal to the ratio of the characteristic rate constants of the reaction. Chemical Reaction Order Order of reaction is refers to the number of molecules involved in forming a reaction complex that is competent to proceed to product(s). Empirically, order is easily determined by summing the exponents of each concentration term in the rate equation for a reaction. A reaction characterized by the conversion of one molecule of A to one molecule of B with no influence from any other reactant or solvent is a first-order reaction. The exponent on the substrate concentration in the rate equation for this type of reaction is 1. A reaction with two substrates forming two products would a second-order reaction. However, the reactants in second- and higher- order reactions need not be different chemical species. An example of a second order reaction is the formation of ATP through the condensation of ADP with orthophosphate: ADP + H2PO4 FIGURE 10. 1 FIGURE 10. 2 For this reaction the forward reaction rate would be written as: ATP + H2O---------8
V forward = k1 [ADP][H2PO4]---------------9 L. Michaelis and M. Menten in 1913 postulated that enzyme first combine reversibly with the substrate to form an enzyme substrate complex in a relatively fast reversible step and then product is released along with free enzyme. As we know at fixed enzyme concentration, when substrate conc. Is increased then initially rate become faster and at maximum velocity where enzyme saturates the rate become steady as shown in figure. The Michaelis derived an equation to explain the fact as Initial velocity Michaelis-Menton constant In typical enzyme-catalyzed reactions, reactant and product concentrations are usually hundreds or thousands of times greater than the enzyme concentration. Consequently, each enzyme molecule catalyzes the conversion to product of many reactant molecules. In biochemical reactions, reactants are commonly known as substrates. The catalytic event that converts substrate to product involves the formation of a transition state, and it occurs most easily at a specific binding site on the enzyme. This site, called the catalytic site of the enzyme, has been evolutionarily structured to provide specific, high-affinity binding of substrate(s) and to provide an environment that favors the catalytic events. The complex that forms when substrate(s) and enzyme combine is called the enzyme substrate (ES) complex. Reaction products arise when the ES complex breaks down releasing free enzyme. Between the binding of substrate to enzyme, and the reappearance of free enzyme and product, a series of complex events must take place. At a minimum S, an ES complex must be formed; this complex must pass to the transition state (ES*); and the transition state complex must advance to an enzyme product complex (EP). The latter is finally competent to dissociate into product and free enzyme. The series of events can be shown thus: E + S ES P-------------------10 and E+S K1 ES ES* EP E +
11
k-1 The ES complex then breaks down in a slower second step to yield the free enzyme and the reaction product P: k2 ES -------- E+P ..(12) Since the second step is slower and therefore it limits the rate of overall reaction. It means that, overall rate of enzyme-catalyzed reaction must be proportional to the concentration of species that reacts in the second step, which is ES. At any time the enzyme-catalyzed reaction, the enzyme exists in the combined form E or uncombined form ES. At low [S], most of the enzyme will be in uncombined form E. here the rate will be proportional to the [s] because the equilibrium of equation will shift for the formation of more ES as S is increased. The maximum initial rate is observed when the entire enzyme E0 is present as ES complex and the concentration of E is very small. Under these state the enzyme is saturated with its substrate, so that further increase in [S] have no effect on rate. This is the condition when [S] is sufficiently high that essentially all the free
enzyme is converted into the ES form. After the ES complex breaks down to yield product P the enzyme is free to catalyze another reaction. a. Pre-steady state:-The duration in which ES formation takes place is called as pre-steady state. It is too difficult to observe because of very short time. b. Steady state:-Here ES complex is constant over a period of time. Derivation: E+S k-1 k1 k2 ES E+P (13)
VO can be determined by the breakdown of ES to give product, which is determined by the [ES]: V0=k2 [ES]-----------------------------------------------------(14) TOTAL enzyme concentration can be represented by [Et]. So free enzyme can be represented by [Et][ES]. Also, because [S] is greater than Et, the amount of substrate bound by the enzyme at any given time is negligible compared to total [S]. .. (15) K1 denotes the rate of formation of ES= k1( [Et][ES] [S]
k-1 denotes the rate of ES breakdown= k-1[ES] + k2 [ES]-------------------------(16) So in the steady state, eq. 5=6 k1( [Et][ES] [S] [ES]-----------------------(17) or k1[Et][S]--- K1[ES] [S] [ES]----------------------------(18) = = k-1[ES] + k2
(k-1 + k2)
To Simplify the equation add the term k1[E] [S] to both side, we finally get K1 [Et][S] k2)[ES]-------------------------(19) Solving for the ES we get. [ES] = --------------------------------(20) or [ES] = --------------------------------(21) [Et][S] -----------------[S] +( k-1+ k2)/ k1 K1[Et][S] = (k1 [S]+k-1+
-----------------k1[S] + k-1+ k2
The term ( k-1+ k2)/ k1 is called as MichaelisMenten equation, the rate equation of one substrate, enzyme catalyzed reaction and is denoted by the Km. this defines the rate of initial velocity and maximum velocity. Putting value of ES in equation 21 from equation 13 gives new final equation that is Vo= V max . [S] / Km + [S]-------------------22
What will happen if the initial velocity is exactly one-half of maximum velocity?
So if V0 = Vmax/2, then Vmax/2= V max [S]/ km+[S],-----------------23 = [S]/ km+[S] solving for km, we get Km+[S] = 2[S], or --------------------24 This represent that Km is equivalent to the substrate concentration at which Vo is one half Vmax. Note that Km has unit of molarity. This shows that the MichaelisMenten constant equals the substrate concentration at half-maximal reaction velocity. Consequently, the dimension of kM is Mol. The smaller the value of kM , the higher is the enzyme's affinity for its substrate. See figure 10.3. Km is (roughly) an inverse measure of the affinity or strength of binding between the enzyme and its substrate. The lower the Km, the greater the affinity (so the lower the concentration of substrate needed to achieve a given rate). Note :for the Michaelis-Menten reaction, k2 is the rate limiting ; thus k2<<k-1 So Km reduces to Km=k-1/k1, and it is defined as dissociation constant, Ks for the ES complex. 1. Km can be very complex in the multistep enzyme catalysis. 2. Km can change from one enzyme to other. 3. Km denotes the affinity of enzyme, but it does not tell anything about the nature of enzyme. 4. An enzyme that acts on very low concentration of substrate has very low Km. high value of Km denotes low enzyme activity. 5. Low Km enzyme can efficiently covert very low amount of substrate into the product such enzyme is said to have high affinity of enzyme while, other enzyme that requires large amount of substances for their activation and conversion into product is said to have as High Km value but low affinity. This property is efficiently utilized by the LDH enzyme that has different Km value in the heart and muscle. In muscle LDH have High Km, while in the heart it has low Km essential for the survival. On writing the Michaelis Menten constant in reciprocal form 1 / v = (kM / vmax) x 1 / [S]--------------------25 This representation is also known as the Lineweaver-Burk equation. Its advantage is in the favourable graphic depiction of the quantities. A double recessive depiction yields a straight line with a gradient of kM / vmax. This plot has advantage that it allows more accurate determination of Vmax. See figure 10.4 & 10.5 1 / kM and 1 / vmax are the points of intersection with the co-ordinates. FIGURE 10. 3 showing rate of reaction and Km at Vmax Figure 10. 4 Graphical method to find the value of Km by plotting the graph between the 1/Vo verses 1/[S] 10.4 Catalytic efficiency and Turnover number. Catalytic efficiency is denoted by the ratio of Kcat/ Km. K cat is the turn over number of enzyme . See from the table below the enzyme having very high value of Kcat have high capacity to convert substrate into the product The turnover number is a measure of catalytic activity. The kcat is a direct measure of the catalytic production of product under saturating substrate conditions.l kcat, the turnover number, is the maximum number of substrate molecules converted to product per
enzyme molecule per unit of time. According to M-M model, k cat = Vmax /Et. Values of k cat range from less than 1/sec to many millions per sec. in multistep enzymatic reaction, one step may be rate limiting that rate limiting step is equivalent to Kcat. Equation 22 can be written as by placing the value of Vmax = Kcat x Et -------------------26 figure 10. 5 showing turn over number of different enzyme. Note that high the TON of an enzyme, best is the enzyme for maximum conversion of substrate into the product. FIGURE 10. 6 The constant Kcat has unit S-1, reciprocal of time. Note that two enzymes catalyzing different reaction may have the same Kcat value (turnover number) yet the rate may be different. It also denotes about the reaction between the enzyme and substrate since at low K cat, Km value will also be unsatisfactory. From equation 26, Vo depends on both Et and S, therefore K cat /Km is a second order rate constant. Therefore Kcat /Km is the best way to compare the catalytic efficiency of the enzyme. Many enzymes have K cat /Km value in the range 108 to 109 . FIGURE 10. 7 The kinetics of simple reactions like that above was first characterized by biochemists Michaelis and Menten. The concepts underlying their analysis of enzyme kinetics continue to provide the cornerstone for understanding metabolism today, and for the development and clinical use of drugs aimed at selectively altering rate constants and interfering with the progress of disease states.
ENZYME KINETICS We start with a very simple expression for formation of the enzyme-substrate complex, ES, from free enzyme, E, and free substrate, S. The rate constants for formation and breakdown back to reactants are k+1 and k-1, respectively. The rate constant for formation of the product is k+2, and to further simplify our treatment, we wil work within the approximation that we can neglect the backreaction, k-2. We can therefore write a simple kinetic scheme as follows: We will work within an approximation called the steady-state approximation, in which the rate of formation of the enzyme-substrate complex is equal to its rate of decay, and is therefore zero, i.e., It is also useful to write down two other definitions and assumptions before we continue: 1. The first thing we need to realize is that the total concentration of enzyme in all of its forms should be equal to the original free enzyme concentration: 2. The second point is that the rate of the reaction should be equal to formation of products, so the initial velocity, vo, and maximum possible velocity, or Vmax, obtained when all the enzyme is substrate-bound, can be written as
Now, before making the steady-state approximation, we can look at the kinetic scheme above and deduce that the rate of change in the concentration of the enzyme-substrate complex should be equal to the rate of formation of the complex minus the rate of decay of the complex. The rate of formation is dependent upon the rate constant k+2 and the enzyme and substrate complexes. The rate of decay involves two possible pathways. One is to form products, and the other is to reform the reactants. Therefore the rate of decay of the enzyme-substrate complex will be the sum of two rate constants, k+2 and k-1. Therefore we can write and from the steady-state approximation, this has to equal zero. Before making this approximation, that equation is insoluble. Now, however, you can solve it fairly simply (despite what I did in class). First, if the steady-state approximation holds, then you can rearrange the equation to look like this: Now group all of the rate constants together We can define this collection of rate constants as something called the Michaelis Constant, or Km. Note that this is not an equilibrium constant. Now we have an even simpler expression: We are almost done. However, we want an expression in terms of constants and measurable quantities such as the substrate concentration, [S], and the initial velocity, vo. To get the equation into that form, we need two things from above. The first is our statement that the total enzyme concentration is equal to the concentration of enzyme in all of its forms, which in turn is the same as the starting concentration of enzyme, i.e.,E total = Eo = [E] + [ES] or conveniently [E] = [Eo] [ES] more, . Then we can substitute that into our previously simplified equation to obtain Solve for ES Since initial velocity V0 = K2 [ES] and max velocity Vmax = K2 [E total]
Chapter-6 Factor affecting the rate of catalysis. 1234Substrate concentration. Temperature pH Inhibitors
The rate at which an enzyme works is influenced by several factors. 1. Concentration of substrate molecules (the more of them available, the quicker the enzyme molecules collide and bind with them). The concentration of substrate is designated [S] and is expressed in unit of molarity.
2. The temperature. As the temperature rises, molecular motion - and hence collisions between enzyme and substrate - speed up. But as enzymes are proteins, there is an upper limit beyond which the enzyme becomes denatured and ineffective. 3. The presence of inhibitors. The study of the rate at which an enzyme works is called enzyme kinetics. Many toxic substances owe their toxic properties to their ability to act as inhibitors (slow down the rate of reactions by different mechanism) to important enzymes responsible for catalyzing important biochemical processes. Once the enzyme is inhibited the process cannot take place, and a toxicological symptom occurs that often leads to paralysis, coma or even death of the organism. For example, cyanide poisoning is due to the cyanide ion competitively inhibiting the active site of the cytochromases enzymes responsible for catalyzing the Oxidation and Reduction processes of the Electron Transport System which is responsible for cellular respiration. Competitive Inhibition occurs when a molecule that is close enough to the shape of the true substrate will fit into the active site. Once locked into position, the blocker molecule prevents the true substrate molecule from getting into position. This effectively blocks the active site. The molecule competes for the active site with the true substrate molecule which is concentration dependent. If the concentration of substrate becomes more than the inhibitor they may replace the inhibitor later on. This is the reason why ethyl alcohol is given to person that has consumed the toxic alcohol (methyl alcohol) so that methyl alcohol can be competitively replaced by ethyl alcohol. Other inhibitors latch themselves not to the active site itself but to some portion of the enzyme molecule close to the active site which results in the changing of the shape of the active site. This is referred to as non-competitive inhibition or mixed inhibition. Many heavy metals like Lead, Mercury, and Chromium will function as non-competitive inhibitors. Toxicology is the study of how toxicological substances can interfere with life sustaining enzymes via inhibition. The pesticide and herbicide industries make use of competitive and Non-Competitive Inhibitors. Biological warfare owes its success to enzyme inhibition but so does the life giving chemotherapeutic treatment of cancerous tumor growths with agents that inhibit important cancel cell enzymes. All in all the use of inhibitors can be used for the benefit of mankind or its destruction. 10.4 .1 Substrate concentration As we know that substrate react with the enzyme and it is converted into the product. If the substrate concentration is, low enough then the graph between the velocity of reaction and substrate utilization, will give almost a linear graph. But if the substrate concentration is high enough then we will obtain the graph that will be parabolic for some time and then straight in term of velocity. See the graph 10.9. FIGURE 10. 8 FIGURE 10. 9 10.4.2 Effect of temperature and pressure Every enzyme has a temperature range of optimum activity. Outside that temperature range the enzyme is rendered inactive and is said to be totally inhibited. This occurs because as the temperature changes this supplies enough energy to break some of the intramolecular attractions between polar groups (Hydrogen bonding, dipole-dipole attractions) as well as the Hydrophobic forces between non-polar groups within the protein structure. When these forces are disturbed and changed, this causes a change in the secondary and tertiary levels of protein structure, and the active site is altered in its conformation beyond its ability to accommodate the substrate molecules it was intended to catalyze. Most enzymes (and
there are hundreds within the human organism) within the human cells will shut down at a body temperature below a certain value which varies according to each individual. This can happen if body temperature gets too low (hypothermia) or too high (hyperthermia). Temperature is an important factor in the regulation of enzyme activity. At some temperature activity becomes zero and at some temperature enzyme efficiency of conversion of substrate into product becomes high. Of all most important factor is the active site, because active site contains amino acid residues inside the pocket, and the ability of these residue to interact with the substrate functional group and capacity to breaking and making bonds is certainly going to be effected by the . Rates of all reactions, including those catalyzed by enzymes, rise with increase in temperature in accordance with the Arrhenius equation. K = A e G*/RT --------------27 OR log K = log A EA /2.3 RT where k is the kinetic rate constant for the reaction, A is the Arrhenius constant, also known as the frequency factor, G* =EA is the standard free energy of activation (kJ M-1) which depends on entropic and enthalpy factors, R is the gas law constant and T is the absolute temperature. Typical standard free energies of activation (15 - 70 kJ M-1) give rise to increases in rate by factors between 1.2 and 2.5 for every 10C rise in temperature. This factor for the increase in the rate of reaction for every 10C rise in temperature is commonly denoted by the term Q10 (i.e. in this case, Q10 is within the range 1.2 - 2.5). All the rate constants contributing to the catalytic mechanism will vary independently, causing changes in both Km and Vmax. It follows that, in an exothermic reaction, the reverse reaction (having higher activation energy) increases more rapidly with temperature than the forward reaction. This, not only alters the equilibrium constant, but also reduces the optimum temperature for maximum conversion as the reaction progresses. The reverse holds for endothermic reactions such as that of glucose isomerase where the ratio of fructose to glucose, at equilibrium, increases from 1.00 at 55C to 1.17 at 80C. In general, it would be preferable to use enzymes at high temperatures in order to make use of this increased rate of reaction plus the protection it affords against microbial contamination. Enzymes, however, are proteins and undergo essentially irreversible denaturation (i.e.. conformational alteration entailing a loss of biological activity) at temperatures above those to which they are ordinarily exposed in their natural environment. These denaturing reactions have standard free energies of activation of about 200 - 300 kJ mole-1 (Q10 in the range 6 - 36) which means that, above a critical temperature, there is a rapid rate of loss of activity (Figure 8.5). The actual loss of activity is the product of this rate and the duration of incubation (Figure 8.6). It may be due to covalent changes such as the deamination of asparagine residues or non-covalent changes such as the rearrangement of the protein chain. Inactivation by heat denaturation has a profound effect on the enzymes productivity (Figure 8.7). ________________________________________ FIGURE 10. 10 FIGURE 10. 11 A schematic diagram showing the effect of the temperature on the activity of an enzyme catalyzed reaction. short incubation period; ----- long incubation period. Note that the temperature at which there appears to be maximum activity varies with the incubation time. ________________________________________ ________________________________________ FIGURE 10. 12
A schematic diagram showing the effect of the temperature on the productivity of an enzyme catalyzed reaction. 55C; 60C; 65C. The optimum productivity is seen to vary with the process time, which may be determined by other additional factors (e.g. overhead costs). It is often difficult to get precise control of the temperature of an enzyme catalyzed process and, under these circumstances, it may be seen that it is prudent to err on the low temperature side. The thermal denaturation of an enzyme may be modeled by the following serial deactivation scheme: --------27.1 Where kd1 and kd2 are the first-order deactivation rate coefficients, E is the native enzyme which may, or may not, be an equilibrium mixture of a number of species, distinct in structure or activity, and E1 and E2 are enzyme molecules of average specific activity relative to E of A1 and A2. A1 may be greater or less than unity (i.e. E1 may have higher or lower activity than E) whereas A2 is normally very small or zero. This model allows for the rare cases involving free enzyme (e.g. tyrosinase) and the somewhat commoner cases involving immobilised enzyme where there is a small initial activation or period of grace involving negligible discernible loss of activity during short incubation periods but prior to later deactivation. Assuming, at the beginning of the reaction: [E] = [E]0 -----------28 and: [E1] = [E2] = 0 ----29 At time t, [E] = [E1] = [E2] = [E0] ----------30 It follows from the reaction scheme [27.1], - d[E] / dt =Kd1[E] -----------------31 Integrating equation 31 using the boundary condition in equation 28 gives: [E] = [E]0 e (-Kdt) = ---------------32 From the reaction scheme - d[E1] / dt =Kd2[E1]-Kd1 [E] ---------------33 Substituting for [E] from equation 32 - d[E1] / dt =Kd2[E1]-Kd1 [E]0 e (-Kdt) -------------34 Integrating equation 33 using the boundary condition in equation 29 gives: [E1]=Kd1 [E]0 / Kd2-Kd1 = (e (-Kd1t) - e (-Kd2t) ) -----------35 If the term 'fractional activity' (Af ) is introduced where, Af = [E] + A1 [E1] + A2 [E2] /[E0]---------36 then, substituting for [E2] from equation 30, gives: Af = [E] + A1 [E1] + A2 ([E0]-[E]-[E1]) /[E0]-------------37 therefore: ------38 When both A1 and A2 are zero, the simple first order deactivation rate expression results Af = e (-Kdt) -----------39 The half-life (t1/2) of an enzyme is the time it takes for the activity to reduce to a half of the original activity (i,e. Af = 0.5). If the enzyme inactivation obeys equation 39, the half-life may be simply derived, Ln (1/2) = =Kd1 t 1/2 -----------------------40 Therefore, t1/2 = 0.693 / Kd1 --------------41 In this simple case, the half-life of the enzyme is shown to be inversely proportional to the rate of denaturation. Many enzyme preparations, both free and immobilised, appear to follow this seriestype deactivation scheme. However because reliable and reproducible data is difficult to obtain, inactivation data should, in general, be assumed to be rather error-prone. It is not surprising that such data can be made to fit a model involving four determined parameters (A1, A2, kd1 and kd2). Despite this possible
reservation, equations 38 and 39 remain quite useful and the theory possesses the definite advantage of simplicity. In some cases the series-type deactivation may be due to structural micro heterogeneity, where the enzyme preparation consists of a heterogeneous mixture of a large number of closely related structural forms. These may have been formed during the past history of the enzyme during preparation and storage due to a number of minor reactions, such as de amidation of one or two asparagine or glutamine residues, limited proteolysis or disulphide interchange. Alternatively it may be due to quaternary structure equilibrium or the presence of distinct genetic variants. In any case, the larger the variability the more apparent will be the series-type inactivation kinetics. The practical effect of this is that usually kd1 is apparently much larger than kd2 and A1 is less than unity. In order to minimise loss of activity on storage, even moderate temperatures should be avoided. Most enzymes are stable for months if refrigerated (0 - 4C). Cooling below 0C in the presence of additives (e.g. glycerol), which prevent freezing, can generally increase this storage stability even further. Freezing enzyme solutions is best avoided as it often causes denaturation due to the stress and pH variation caused by ice-crystal formation. The first order deactivation constants are often significantly lower in the case of enzyme-substrate, enzymeinhibitor and enzyme-product complexes which helps to explain the substantial stabilizing effects of suitable ligand, especially at concentrations where little free enzyme exists (e.g. [S] >> Km). Other factors, such as the presence of thiols anti-oxidants, may improve the thermal stability in particular cases. It has been found that the heat denaturation of enzymes is primarily due to the proteins' interactions with the aqueous environment. They are generally more stable in concentrated, rather than dilute, solutions. In a dry or predominantly dehydrated state, they remain active for considerable periods even at temperatures above 100C. This property has great technological significance and is currently being exploited by the use of organic solvents. Pressure changes will also affect enzyme catalyzed reactions. Clearly any reaction involving dissolved gases (e.g. oxygenases and decarboxylases) will be particularly affected by the increased gas solubility at high pressures. The equilibrium position of the reaction will also be shifted due to any difference in molar volumes between the reactants and products. However an additional, if rather small, influence is due to the volume changes which occur during enzymic binding and catalysis. Some enzyme-reactant mixtures may undergo reductions in volume amounting to up to 50 ml mole-1 during reaction due to conformational restrictions and changes in their hydration. This, in turn, may lead to a doubling of the kcat, and/or a halving in the Km for a 1000 fold increase in pressure. The relative effects on kcat and Km depend upon the relative volume changes during binding and the formation of the reaction transition states. 10.4.3 Effect of pH on Enzyme Enzymes are amphoteric molecules containing a large number of acid and basic groups, mainly situated on their surface. The charges on these groups will vary, according to their acid dissociation constants, with the pH of their environment (Table 1.1). This will affect the total net charge of the enzymes and the distribution of charge on their exterior surfaces, in addition to the reactivity of the catalytically active groups. These effects are especially important in the neighborhood of the active sites. Taken together, the changes in charges with pH affect the activity, structural stability and solubility of the enzyme. Therefore all enzymes are affected by the change of pH. They act slowly on the low pH or high pH. So an optimum pH is needed for its full activity. Figure 10. 13 Figure showing optimum pH for two separate enzymes. This is because of change in the ionization pattern of the amino acid residue inside the active site of the enzyme. Either proton is removed or added. Changes in the pH or acidity of the environment can take place that would alter or totally inhibit the enzyme from catalyzing a reaction. This change in the pH will affect
the polar and non-polar intramolecular attractive and repulsive forces and alter the shape of the enzyme and the active site as well to the point where the substrate molecule could no longer fit, and the chemical change would be inhibited from taking place as efficiently or not at all. In an acid solution any basic groups such as the Nitrogen groups in the protein would be protonated. If the environment was too basic the acid groups would be deprotonated. This would alter the electrical attractions between polar groups. Every enzyme has an optimum pH range outside of which the enzyme is inhibited. Some enzymes like many of the hydrolytic enzymes in the stomach such as Pepsin and Chymotrypsin effective operate at a very low acidic pH. Other enzymes like alpha amylase found in the saliva of the mouth operate most effectively at near neutrality. Still other enzymes like the lipases will function most effectively at basic pH values. If the pH drops in the blood called acidosis then enzymes in the blood will be inhibited outside their optimal pH range. If the pH climbs to an unacceptably high value called alkalosis then enzymes ceases to function effectively. Normally, these conditions do not take place because of the highly efficient buffers found in the blood that restrict the pH of the blood to a very narrow range. Buffers are a substance or mixtures of substances that resist any change in the pH. There are many buffer systems found in the body to adjust the pH so that enzymes might continue to catalyze their reactions. Correcting pH or temperature imbalances will usually allow the enzyme to resume its original shape or conformation. Some substances when added to the system will irreversibly break bonds disrupting the primary structure so that the enzyme is inhibited permanently. The enzyme is said to be irreversably denatured. Many toxic substances will break covalent bonds and cause the unraveling of the protein enzyme. Other toxic substances will precipitate enzymes effectively removing them from the solution thus preventing them from catalyzing the reaction. This is also called denaturation. Table II pH for Optimum Activity Enzyme pH Optimum Lipase (pancreas) 8.0 Lipase (stomach) 4.0 - 5.0 Lipase (castor oil) 4.7 Pepsin 1.5 - 1.6 Trypsin 7.8 - 8.7 Urease 7.0 Invertase 4.5 Maltase 6.1 - 6.8 Amylase (pancreas) 6.7 - 7.0 Amylase (malt) 4.6 - 5.2 Catalase 7.0 There will be a pH, characteristic of each enzyme, at which the net charge on the molecule is zero. This is called the isoelectric point (pI), at which the enzyme generally has minimum solubility in aqueous solutions. In a similar manner to the effect on enzymes, the charge and charge distribution on the substrate(s), product(s) and coenzymes (where applicable) will also be affected by pH changes. Increasing hydrogen ion concentration will, additionally, increase the successful competition of hydrogen ions for any metal cationic binding sites on the enzyme, reducing the bound metal cation concentration. Decreasing hydrogen ion concentration, on the other hand, leads to increasing hydroxyl ion concentration which competes against the enzymes' ligand for divalent and trivalent cations causing their conversion to hydroxides and, at high hydroxyl concentrations, their complete removal from the enzyme. The temperature also has a marked effect on ionizations, the extent of which depends on the heats of ionization of the particular groups concerned. The relationship between the change in the pKa and the change in temperature is given by a derivative of the Gibbs-Helmholtz equation.
where T is the absolute temperature (K), R is the gas law constant (8.314 J M-1 K1), H is the heat of ionisation and the numeric constant (2.303) is the natural logarithm of 10, as pKa's are based on logarithms with base 10. This variation is sufficient to shift the pI of enzymes by up to one unit towards lower pH on increasing the temperature by 50C. These charge variations, plus any consequent structural alterations, may be reflected in changes in the binding of the substrate, the catalytic efficiency and the amount of active enzyme. Both Vmax and Km will be affected due to the resultant modifications to the kinetic rate constants k+1, k-1 and kcat (k+2 in the Michaelis-Menten mechanism), and the variation in the concentration of active enzyme. The effect of pH on the Vmax of an enzyme catalysed reaction may be explained using the, generally true, assumption that only one charged form of the enzyme is optimally catalytic and therefore the maximum concentration of the enzyme-substrate intermediate cannot be greater than the concentration of this species. In simple terms, assume EH- is the only active form of the enzyme, The variation of activity with pH, within a range of 2-3 units each side of the pI, is normally a reversible process. Extremes of pH will, however, cause a timeand temperature-dependent, essentially irreversible, denaturation. In alkaline solution (pH > 8), there may be partial destruction of cystine residues due to base catalysed b-elimination reactions whereas, in acid solutions (pH < 4), hydrolysis of the labile peptide bonds, sometimes found next to aspartic acid residues, may occur. The importance of the knowledge concerning the variation of activity with pH cannot be over-emphasized. However, a number of other factors may mean that the optimum pH in the Vmax-pH diagram may not be the pH of choice in a technological process involving enzymes. These include the variation of solubility of substrate(s) and product(s), changes in the position of equilibrium for a reaction, suppression of the ionization of a product to facilitate its partition and recovery into an organic solvent, and the reduction in susceptibility to oxidation or microbial contamination. The major such factor is the effect of pH on enzyme stability. This relationship is further complicated by the variation in the effect of the pH with both the duration of the process and the temperature or temperature-time profile. The important parameter derived from these influences is the productivity of the enzyme (i.e. how much substrate it is capable of converting to product). The variation of productivity with pH may be similar to that of the Vmax-pH relationship but changes in the substrate stream composition and contact time may also make some contribution. Generally, the variation must be determined under the industrial process conditions. It is possible to alter the pH-activity profiles of enzymes. The ionisation of the carboxylic acids involves the separation of the released groups of opposite charge. This process is encouraged within solutions of higher polarity and reduced by less polar solutions. Thus, reducing the dielectric constant of an aqueous solution by the addition of a co-solvent of low polarity (e.g. dioxan, ethanol), or by immobilisation, increases the pKa of carboxylic acid groups. This method is sometimes useful but not generally applicable to enzyme catalysed reactions as it may cause a drastic change on an enzyme's productivity due to denaturation (. The pKa of basic groups are not similarly affected as there is no separation of charges when basic groups ionise. However, protonated basic groups which are stabilised by neighbouring negatively charged groups will be stabilised (i.e. have lowered pKa) by solutions of lower polarity. Changes in the ionic strength (I) of the solution may also have some effect. The ionic strength is defined as half of the total sum of the concentration (ci) of every ionic species (i) in the solution times the square of its charge (zi); i.e. I=0.5(CiZi2) .For example, the ionic strength of a 0.1 M solution of CaCl2 , is 0.5 x (0.1 x 22 + 0.2 x 12) = 0.3 M. At higher solution ionic strength, charge separation is encouraged with a concomitant lowering of the carboxylic acid pKas. These changes, extensive as they may be, have little effect on the overall charge on the enzyme molecule at neutral pH and are, therefore, only likely to exert a small influence on the enzyme's
isoelectric point. Chemical derivatisation methods are available for converting surface charges from positive to negative and vice-versa. It is found that a single change in charge has little effect on the pH-activity profile, unless it is at the active site. However if all lysines are converted to carboxylates (e.g. by reaction with succinic anhydride) or if all the carboxylates are converted to amines (e.g. by coupling to ethylene diamine by means of a carbodiimide the profile can be shifted about a pH unit towards higher or lower pH, respectively. The cause of these shifts is primarily the stabilisation or destabilisation of the charges at the active site during the reaction, and the effects are most noticeable at low ionic strength. The ionic strength of the solution is an important parameter affecting enzyme activity. This is especially noticeable where catalysis depends on the movement of charged molecules relative to each other. Thus both the binding of charged substrates to enzymes and the movement of charged groups within the catalytic 'active' site will be influenced by the ionic composition of the medium. If the charges are opposite then there is a decrease in the reaction rate with increasing ionic strength whereas if the charges are identical, an increase in the reaction rate will occur (e.g. the rate controlling step in the catalytic mechanism of chymotrypsin involves the approach of two positively charged groups, 57histidine+ and 145arginine+ causing a significant increase in kcat on increasing the ionic strength of the solution). Even if a more complex relationship between the rate constants and the ionic strength holds, it is clearly important to control the ionic strength of solutions in parallel with the control of pH.
FIGURE 10. 14 showing molecule activation at high and low temperature Enzyme Substrate Product Rate without Enzyme moles/L per min Rate with Enzyme moles/L per min Acceleration due to Enzyme Hexokinase Glucose Glucose 6-Phosphate <.0000001 1300 > 13 billion Phosphorylase <.000000005 1600 > 320 billion Alcohol Dehydrogenase Ethanol Acetaldehyde <.000006 2700 > 450 million Creatine Kinase Creatine Creatine Phosphate <.003 40 > 13, 000 o From: Daniel Koshland, Jr. Molecular geometry in enzyme action. Journal of Cellular & Comparative Physiology 47: 217-234, 1956.
Chapter-7 Enzyme inhibition A number of substances may cause a reduction in the rate of an enzyme catalysed reaction. Some of these (e.g. urea) are non-specific protein denaturants. Others, which generally act in a fairly specific manner, are known as inhibitors. Loss of activity may be either reversible, where activity may be restored by the removal
of the inhibitor, or irreversible, where the loss of activity is time dependent and cannot be recovered during the timescale of interest. If the inhibited enzyme is totally inactive, irreversible inhibition behaves as a time-dependent loss of enzyme concentration (i.e.. lower Vmax), in other cases, involving incomplete inactivation.There may be time-dependent changes in both Km and Vmax. Heavy metal ions (e.g. mercury and lead) should generally be prevented from coming into contact with enzymes as they usually cause such irreversible inhibition by binding strongly to the amino acid backbone. More important for most enzyme-catalysed processes is the effect of reversible inhibitors. These are generally discussed in terms of a simple extension to the Michaelis-Menten reaction scheme. --------------------42 where I represents the reversible inhibitor and the inhibitory (dissociation) constants Ki and Ki' are given by --------------------43 and, --------------------------44 For the present purposes, it is assumed that neither EI nor ESI may react to form product. Equilibrium between EI and ESI is allowed, but makes no net contribution to the rate equation as it must be equivalent to the equilibrium established through: ------45 Binding of inhibitors may change with the pH of the solution, as discussed earlier for substrate binding, and result in the independent variation of both Ki and Ki' with pH. In order to simplify the analysis substantially, it is necessary that the rate of product formation (k+2) is slow relative to the establishment of the equilibria between the species. Therefore: -----------46 also ----------------------------47 where: -----------48 therefore ---------------49 Substituting from equations (43), ( 44) and (46), followed by simplification, gives: --------------50 therefore --------------51 If the total enzyme concentration is much less than the total inhibitor concentration (i.e. [E]0<< [I]0), then: --------------------52 This is the equation used generally for mixed inhibition involving both EI and ESI complexes (Figure 10.19a). A number of simplified cases exist that are reversible. 1. Competitive inhibition. 2. Noncompetitive inhibition 3. Uncompetitive inhibition. Figure 11. 1 showing effect of concentration of inhibitor on enzyme activity on different time interval. A. Competitive inhibition This occurs when both the substrate and inhibitor compete for binding to the active site of the enzyme. The inhibition is most noticeable at low substrate concentrations but can be overcome at sufficiently high substrate concentrations as the Vmax remains unaffected (Figure 10.19b). Normal Vmax can be observed when substrate is sufficiently present. In the presence of a competitive inhibitor, it
takes a higher substrate concentration to achieve the same velocities that were reached in its absence. So while Vmax can still be reached if sufficient substrate is available, one-half Vmax requires a higher [S] than before and thus Km is larger. But at [S] at which Vo=1/Vmax, the Km (apparent Km) will increase in presence of inhinitor. In double reciprocal plot this can be obsereved. Ki' is much greater than the total inhibitor concentration and the ESI complex is not formed but EI is formed. The rate equation is given by: ---------------------52 where Kmapp is the apparent Km for the reaction, and is given by -----------------53 Normally the competitive inhibitor bears some structural similarity to the substrate, and often is a reaction product (product inhibition, e.g. inhibition of lactase by galactose), which may cause a substantial loss of productivity when high degrees of conversion are required. The rate equation for product inhibition is derived from equations (52) and (53). -----54 Figure 11. 2 figure showing competitive inhibition 1/Vo (1/ M/min) verses 1/[S] (1/ mM). note that on increasing inhibitor concentration 1/Vmax remain unchanged while Km value changes. Figure 11. 3 showing effect of concentration of inhibitor on enzyme activity.
A similar effect is observed with competing substrates, quite a common state of affairs in industrial conversions, and especially relevant to macromolecular hydrolyses where a number of different substrates may coexist, all with different kinetic parameters. The reaction involving two co-substrates may be modelled by the scheme. ---------------55 Both substrates compete for the same catalytic site and, therefore, their binding is mutually exclusive and they behave as competitive inhibitors of each others reactions. If the rates of product formation are much slower than attainment of the equilibria (i.e. k+2 and k+4 are very much less than k-1 and k-3 respectively), the rate of formation of P1 is given by: ---------56 and the rate of formation of P2 is given by ---------------57 If the substrate concentrations are both small relative to their Km values: --------------------58 Therefore, in a competitive situation using the same enzyme and with both substrates at the same concentration: --------------59 Where and > in this simplified case. The relative rates of reaction are in the ratio of their specificity constants. If both reactions produce the same product (e.g. some hydrolyses): ---------------60 therefore --------------------------61 B. Uncompetitive inhibition This occurs when the inhibitor binds to a site which only becomes available after the substrate (S1) has bound to the active site of the enzyme. Ki is much greater than the total inhibitor concentration and the EI complex is not formed. This inhibition is most commonly encountered in multi-substrate reactions where the inhibitor is competitive with respect to one substrate (e.g. S2) but uncompetitive with respect to another (e.g. S1), where the reaction scheme may be represented by. --------61
The inhibition is most noticeable at high substrate concentrations (i.e. S1 in the scheme above) and cannot be overcome as both the Vmax and Km are equally reduced (Figure 1.8c). The rate equation is: ----------------------62 where Vmaxapp and Kmapp are the apparent Vmax and Km given by --------------------63 and --------------------64 Figure 11. 4
Figure 11. 5 figure showing case of uncompetitive inhibition In this case the specificity constant remains unaffected by the inhibition. Normally the uncompetitive inhibitor also bears some structural similarity to one of the substrates and, again, is often a reaction product. ________________________________________ figure 11.6 A schematic diagram showing the effect of reversible inhibitors on the rate of enzyme-catalysed reactions. no inhibition, (a) mixed inhibition ([I] = Ki = 0.5 Ki'); lower Vmaxapp (= 0.67 Vmax), higher Kmapp (= 2 Km). (b) competitive inhibition ([I] = Ki); Vmaxapp unchanged (= Vmax), higher Kmapp (= 2 Km). (c) uncompetitive inhibition ([I] = Ki'); lower Vmaxapp (= 0.5 Vmax) and Kmapp (= 0.5 Km). (d) noncompetitive inhibition ([I] = Ki = Ki'); lower Vmaxapp (= 0.5 Vmax), unchanged Kmapp (= Km). ________________________________________ A special case of uncompetitive inhibition is substrate inhibition which occurs at high substrate concentrations in about 20% of all known enzymes (e.g. invertase is inhibited by sucrose). It is primarily caused by more than one substrate molecule binding to an active site meant for just one, often by different parts of the substrate molecules binding to different sites within the substrate binding site. If the resultant complex is inactive this type of inhibition causes a reduction in the rate of reaction, at high substrate concentrations. It may be modeled by the following scheme ---------------65 where ---------------------66 The assumption is made that ESS may not react to form product. It follows from equation (52) that: -------------------------67 Even quite high values for KS lead to a leveling off of the rate of reaction at high substrate concentrations, and lower KS values cause substantial inhibition (Figure 10.20). ________________________________________ Figure 11. 7 The effect of substrate inhibition on the rate of an enzymecatalysed reaction. A comparison is made between the inhibition caused by increasing KS relative to Km. no inhibition, KS/Km >> 100; KS/Km = 100; KS/Km = 10; KS/Km = 1. By the nature of the binding causing this inhibition, it is unlikely that KS/Km < 1. ________________________________________ C. Noncompetitive inhibition This occurs when the inhibitor binds at a site away from the substrate binding site, causing a reduction in the catalytic rate. The inhibitor binding causes inactivation of enzyme due to which apparent Vmax (Vmax = Kcat [Et] ) lower down whether or not substrate is bound. enzyme rate (velocity) is reduced for all values of [S], including Vmax and one-half Vmax but Km remains unchanged because the active site of those enzyme molecules that have not been inhibited is unchanged Therefore there is no
effect on Km. It is quite rarely found as a special case of mixed inhibition. Both the EI and ESI complexes are formed equally well (i.e. Ki equals Ki'). The fractional inhibition is identical at all substrate concentrations and cannot be overcome by increasing substrate concentration due to the reduction in Vmax . The rate equation is given by: Figure 11. 8 figure showing Noncompetative inhibition slope is Km /Vmax ---------------------------68 where Vmaxapp is given by: --------------------------------69 The diminution in the rate of reaction with pH, described earlier, may be considered as a special case of noncompetitive inhibition; the inhibitor being the hydrogen ion on the acid side of the optimum or the hydroxide ion on the alkaline side. Figure 11. 9
Figure 11. 6 (b) Determination of Vmax and Km It is important to have as thorough knowledge as is possible of the performance characteristics of enzymes, if they are to be used most efficiently. The kinetic parameters Vmax, Km and kcat/Km should, therefore, be determined. There are two approaches to this problem using either the reaction progress curve (integral method) or the initial rates of reaction (differential method). Use of either method depends on prior knowledge of the mechanism for the reaction and, at least approximately, the optimum conditions for the reaction. If the mechanism is known and complex then the data must be reconciled to the appropriate model (hypothesis), usually by use of a computer-aided analysis involving a weighted least-squares fit. Many such computer programs are currently available and, if not, the programming skill involved is usually fairly low. If the mechanism is not known, initial attempts are usually made to fit the data to the Michaelis-Menten kinetic model. ------------------70 which, on integration, using the boundary condition that the product is absent at time zero and by substituting [S] by ([S]0 - [P]), becomes ------------71 If the fractional conversion (X) is introduced, where -----------------------72 then equation (72) may be simplified to give: ----------73 Use of equation (70) involves the determination of the initial rate of reaction over a wide range of substrate concentrations. The initial rates are used, so that [S] = [S]0, the predetermined and accurately known substrate concentration at the start of the reaction. Its use also ensures that there is no effect of reaction reversibility or product inhibition which may affect the integral method based on equation (73). Equation (70) can be utilised directly using a computer program, involving a weighted least-squares fit, where the parameters for determining the hyperbolic relationship between the initial rate of reaction and initial substrate concentration (i.e.. Km and Vmax) are chosen in order to minimise the errors between the data and the model, and the assumption is made that the errors inherent in the practically determined data are normally distributed about their mean (error-free) value. Alternatively the direct linear plot may be used (Figure 10.25). This is a powerful non-parametric statistical method which depends upon the assumption that any errors in the experimentally derived data are as likely to be positive (i.e. too high) as negative (i.e. too low). It is common practice to show the data
obtained by the above statistical methods on one of three linearised plots, derived from equation (70) (Figure 10.25). Of these, the double reciprocal plot is preferred to test for the qualitative correctness of a proposed mechanism, and the Eadie-Hofstee plot is preferred for discovering deviations from linearity. ________________________________________ Figure 11. 7 The direct linear plot. A plot of the initial rate of reaction against the initial substrate concentration also showing the way estimates can be directly made of the Km and Vmax. Every pair of data points may be utilised to give a separate estimate of these parameters (i.e. n(n-1)/2 estimates from n data points with differing [S]0). These estimates are determined from the intersections of lines passing through the (x,y) points (-[S]0,0) and (0,v); each intersection forming a separate estimate of Km and Vmax. The intersections are separately ranked in order of increasing value of both Km and Vmax and the median values taken as the best estimates for these parameters. The error in these estimates can be simply determined from sub-ranges of these estimates, the width of the subrange dependent on the accuracy required for the error and the number of data points in the analysis. In this example there are 7 data points and, therefore, 21 estimates for both Km and Vmax. The ranked list of the estimates for Km (mM) is 0.98,1.65, 1.68, 1.70, 1.85, 1.87, 1.89, 1.91, 1.94, 1.96, 1.98, 1.99, 2.03, 2.06, 1.12, 2.16, 2.21, 2.25, 2.38, 2.40, 2.81, with a median value of 1.98 mM. The Km must lie between the 4th (1.70 mM) and 18th (2.25 mM) estimate at a confidence level of 97% (Cornish-Bowden et al., 1978). The list of the estimates for Vmax ( M.min-1) is ranked separately as 3.45, 3.59, 3.80, 3.85, 3.87, 3.89, 3.91, 3.94, 3.96, 3.96, 3.98, 4.01, 4.03, 4.05, 4.13, 4.14, 4.18, 4.26, 4.29, 4.35, with a median value of 3.98 M.min-1. The Vmax must lie between the 4th (3.85 M.min-1) and 18th (4.18 M.min-1) estimate at a confidence level of 97%. It can be seen that outlying estimates have little or no influence on the results. This is a major advantage over the least-squared statistical procedures where rogue data points cause heavily biased effects. ________________________________________ Three ways in which the hyperbolic relationship between the initial rate of reaction and the initial substrate concentration can be rearranged to give linear plots (a) Lineweaver-Burk (double-reciprocal) plot of 1/v against 1/[S]0 giving intercepts at 1/Vmax and -1/Km --------------74 Figure 11. 8 (a) Lineweaver-Burk (double-reciprocal) plot of 1/v against 1/[S]0 giving intercepts at 1/Vmax and -1/Km Figure 11. 9 (b) Eadie-Hofstee plot of v against v/[S]0 giving intercepts at Vmax and Vmax/Km (75) Figure 11. 10 c Hanes-Woolf (half-reciprocal) plot of [S]0/v against [S]0 giving intercepts at Km/Vmax and Km. ----------76 The progress curve of the reaction (Figure 10.28) can be used to determine the specificity constant (kcat/Km) by making use of the relationship between time of reaction and fractional conversion (see equation (73). This has the advantage over the use of the initial rates (above) in that fewer determinations need to be made, possibly only one progress curve is necessary, and sometimes the initial rate of reaction is rather difficult to determine due to its rapid decline. If only the early part of the progress curve, or its derivative, is utilised in the analysis, this procedure may even be used in cases where there is competitive inhibition by
the product, or where the reaction is reversible. ________________________________________ Figure 11. 11 FIGURE 10. 15 . A schematic plot showing the amount of product formed (productivity) against the time of reaction, in a closed system. The specificity constant may be determined by a weighted least-squared fit of the data to the relationship given by equation (73). Table -2
Module III Enzyme Kinetics: Single substrate steady state kinetics; Michaelis Menten equation, Linear plots, King-Altmans method; Inhibitors and activators; Multisubstrate systems; ping-pong mechanism, Alberty equation, Sigmoidal kinetics and Allosteric enzymes Chapter-8 Multisubstrate enzymes Many Enzymes react with two or more substrates simultaneously (includes cofactors : ATP, NADPH, NADP, FADH, FAD etc) Michaelis Menten kinetics is observed when only one substrate is varied with the other(s) held constant (usually the cofactors) Common observed Mechanisms: Sequential All substrates are bound by enzyme before the any products are formed and released Ordered : Substrates bind and products released in specific order Random : no order Ping Pong When one or more product(s) are released before all substrates are bound by enzyme (acyl/phosphoryl enzyme intermediate)
sequential means all substrates on before any products off, while ping pong has products off before all substrates on - can be ordered or random - e.g.: ________________________________________ Adenylate kinase displays sequential kinetics, in which both substrates must be bound before any product is released. This is distinguished from what is termed ping-pong kinetics, in which one reactant modifies the enzyme, and then a second reactant interacts with the modification. Sequential kinetics can be distinguished from ping-pong kinetics by initial rate studies. In practice, measure initial rates as a function of one substrate while holding the other constant. Then, vary the concentration of the second substrate and repeat. Lineweaver-Burk (double-reciprocal) analysis should yield a family of lines that intersect at the left of the y-axis of the graph. Within the realm of sequential reactions lies ordered sequential and random sequential at the extreme ends. In ordered sequential reactions, one substrate is obligated to bind to the enzyme before a second substrate. In random sequential mechanisms there is no preference. In practice, there is usually some degree of order in binding.
DG= refers to the difference in free energy between the transition state and the substrate Enzymes work by decreasing DG= to facilitate more rapid formation of P Think of it as enzymes helping the chemical reaction get over the hump in order to form product (P) ES complex demonstrated in a variety of ways including X-ray crystallography, electron microscopy, and spectrophotometry Enzymes derive much of their catalytic power by bringing in a substrate molecule at a favorable orientation (this is how enzymes reduce the free energy required to convert S into P) Leonor Michaelis (1913): noticed that at constant [enzyme] the rate of a reaction increases with increasing [S] until a maximal velocity (Vmax) is achieved This saturation effect is an important distinction versus uncatalyzed reactions Interpretation of data: ES complexes formed until substrate saturation occurs at which point no more substrate binding sites (i.e., enzyme molecules) are available The kinetic mechanism of an enzyme is simply the sequence in which substrates bind to, and products are released from the enzyme. It should not be confused with the chemical mechanism which is concerned with the chemical interactions between the enzyme and its substrate(s) which results in the creation of product(s). Kinetic mechanisms can be broadly divided into two main types, sequential and ping-pong (also known as double displacement) mechanisms. Sequential mechanisms Sequential reactions are ones in which all reactants bind to the enzyme
before the first product is released. They can be further subdivided into ordered reactions, in which the reactants and products are bound and released in an obligatory sequence, and random reactions, in which there is no obligatory binding sequence. Ping-pong mechanisms Ping-pong reactions are ones in which at least one product is released before all the substrates have bound. To clarify these distinctions we'll look at each of these mechanisms in turn using a typical bi bi enzyme: A + B P + Q We'll start by looking at an ordered sequential reaction, which is perhaps the simplest in kinetic terms. Types of specificity: o Geometric specificity: shape Chiral specificity: most chirally specific enzymes are absolutely stereospecific. Prochirality, because of their own chiral nature enzymes can often hold substrates in such a way that on one chiral product is made, distinguishing between seemingly identical groups. o Chemical specificity: functional groups, types of chemical reaction. Enzyme Catalysis We will look at catalysis in two types of systems: Model systems in organic chemistry to elucidate probable mechanisms of chemical catalysis. Example enzymes to demonstrate these mechanisms in enzyme catalysis. Mechanisms of Chemical Catalysis Look at some examples of catalysis in model systems (organic chemistry) and how they might operate in enzymes. Types of Catalysis: Acid/Base o Specific (H+ & OH- in water) o General (Bronsted acid definition: proton donor/acceptor pairs; buffers.) Covalent o nucleophilic o electrophilic Proximity/orientation Stabilization of Transition State Conformation (Strain/distortion; Charge neutralization) Metal/Metal ion. So how does catalysis work? Recall that the slow step of a reaction is reaching the transition state. Thus if we can find a way to stabilize the transition state (lower Ea) then the reaction rate will be enhanced. Generally we will be looking at three ways to increase rates 1. stabilize transition states 2. increase the concentrations of intermediates 3. use a different reaction pathway. o Plots of vi = d[P]/dt vs. [S] for 0 - 3rd order First Order: r = k[S] for S P; & r =-d[S]/dt = d[P]/dt. o Example - SN1 from organic chemistry: A + B C + D as 1st order reaction, as in the hydroxylation of t-alkylchloride: First order because rate depends only on the formation of the carbocation, which in turn depends only on [R3CCl] Second order: r = k[A][B] for A + B P; or can have r = k[A]2 ; o Example - SN2 from organic chemistry: A + B C + D as 2nd order reaction, as in the hydroxylation of primary alkylchlorides:
Now 2nd order because both RH2CCl and OH- (reverse of figure above) are involved in slow step. Higher order reactions occur, but uncommon. Zero order: r = k: Only occurs with catalysts, important in enzyme catalysis. 0 order also only occurs above a minimum [A]. one-substrate enzymes: For simple enzyme, S P get rectangular hyperbola type plot for vi vs [S] (text Figure 6-11), similar to Mb binding curve. Let's look at a mathematical model and attempt to generate curve. This was first done by Michaelis and Menten for an equilibrium model. Better is the steady state model of Haldane and Briggs (more general), which we will derive. For S P assume And for initial reaction conditions [P] = 0 & therefore k4 = 0, so have Now vi = d[P]/dt = k3[ES] (Note that kcat is often used instead of k3); Assume steady state (steady state assumption: d[ES]/dt= 0): d[ES]/dt= 0; Thus: 0 = d[ES]/dt= k1[E][S] - k2[ES] - k3[ES]. So, Which is known as the Michaelis-Menten Equation. For simple, one-substrate enzymes then, have Michaelis-Menten Equation as a model for enzyme activity. Note predicted consequences of model: [S] >> KM; then vi = Vmax and get Zero order (r = k) [S] << KM; then , and get First order (r = k [S]) [S] = KM; then vi = Vmax/2 This is definition of KM, the substrate concentration at half-saturation. Note consequences for a plot: start off with approximately linear slope with y = kx. Then at the limit of high concentrations have a horizontal line. This is exactly what we expect if we look at the general form of the equation: , the formula for a rectangular hyperbola: (text Figure 6-12) Turnover Number. The rate constant (First order) for the breakdown of the [ES] complex, kcat (k3), is also known as the turnover number, that is the maximum number of substrate molecules processed/active site (moles substrate/mole active site): kcat=Vmax / [E]total. Note that this is best determined under saturating conditions. (text Table 6-08) At very low concentrations of [S] can find the second-order rate constant for the conversion of E + S E + P: vo = (kcat / KM)([E][S]. Linear plots for enzyme kinetic studies Double Reciprocal or Lineweaver-Burke Plot: Need in form: y = ax + b , so take reciprocals of both sides and have . (text Box 6-01 figure 6-1) Other linear plots are also available, and are better in terms of statistics (L-B one of worst, best quality points [high concentration] have least influence on slope, while low precision points [low concentration] are more spread out, and have a large moment, with a strong influence on the slope and the KM intercept this is not as much of a concern now with computer statistical packages, but you still have to understand the statistics). We will see others in the laboratory discussion. FYI - The Eadie-Hofstee Plot One common plot is shown below. Note that the data points are distributed much more evenly over the plot giving better statistics for the slope. In addition the
value of KM is obtained from the slope, giving better precision.
ENZYME KINETICS AND INHIBITION What's exciting about enzyme inhibition? Potential to tell us about enzyme. Potential uses as drugs and toxins. o Understanding drugs and toxins to counter etc. Three major types of inhibition: Competitive inhibition Noncompetitive inhibition Uncompetitive inhibition Competitive Inhibition: S & I are mutually exclusive, E can bind to one OR the other. Plots: (text Box 6-2 figure 1) We can model this inhibition with chemical equations, keeping in mind that S & I are mutually exclusive, E can bind to one OR the other: (text Figure 6-15a) Model: ; and: ; where . Classically assume binding to same site, but other possibilities also. 1. steric hindrance between S & I in different sites. 2. overlapping sites for S & I. 3. Partial sharing of sites. 4. Conformational change of enzyme with binding of either such that other can not bind. Noncompetitive: the inhibitor can bind to either E or ES. S & I do not bind to the same sites! (text Figure 6-15c) Model: ; and . Note that will have two inhibitor binding constants, they may be the same, as in the equation above, or could be different, leading to more complex behavior. Plots for classic, simple situation (overhead MvH 11.5): Mixed: Like non-competitive above, Model: ; and but with different values of Ki. This is in fact the more common situation, with a L-B plot with an intersection on the plot above the 1/vo axis. (text Box 6-2 figure 3) Uncompetitive: In uncompetitive inhibition the inhibitor binds ONLY to the ES complex (text Figure 6-15b). Model: ; and . For double reciprocal plots get parallel lines! (text Box 6-2 figure 2) This is not generally found for single substrate enzymes, but is found in multi-substrate systems. Multi-substrate Enzymes Look at three common and easily understood types. We will use Cleland Nomenclature and "Kinetic mechanism diagrams." Ordered Sequential Bi Bi mechanism (two on; two off); Note: A must bind first, Q is released last. Ping Pong Bi Bi (one on, one off; one on, one off); Note: have some sort of modified enzyme intermediate (often covalent intermediate) Random Sequential Bi Bi (two on; two off); Note: A or B may bind first, P or Q may be released last. Two-substrate Enzyme Product Inhibition Patterns
(Based on: E. B. Cunningham, Biochemistry: Mechanisms of Metabolism. McGraw-Hill Book Company, New York (1978), and W. Cleland, "Substrate Inhibition: in Contemporary Enzyme Kinetics and Mechanism. (Daniel L. Purich, ed.) Academic Press, New york (1983)) Kinetic Mechanism Variable Substrate Product Type of Inhibition Ordered Sequential Bi Bi Ordered Sequential Bi Bi A B Q Noncompetitive A P Noncompetitive B P Noncompetitive Random Sequential Random Sequential B Q A P B P Ping pong Bi Bi Ping pong Bi Bi B Q A P B P Bi Bi Bi Bi A Q Noncompetitive Noncompetitive Noncompetitive Q Competitive
Noncompetitive
A Q Competitive Noncompetitive Noncompetitive Competitive
Note that in each case we can predict/explain the pattern of inhibition on the basis of the substrate and inhibitor binding to the same "enzyme form." Thus for the Ordered Sequential mechanism only the first substrate and last product bind to the same form, in this case the free enzyme. Similarly for the Ping pong mechanism the first substrate and last product should be competitive as the both bind the free enzyme. In this case we also see a competitive inhibition between the second substrate and the first product, since they both bind to the E-X complex. The Random Sequential mechanism is a bit more subtle. Here we see across the board noncompetitive since in each case the substrates (and products) can each bind to more than one substrate form, so competitive inhibition will not be possible! (Think of the product as competing with one order of binding but not the other.) The ping-pong (double displacement) mechanism ________________________________________ The distinguishing feature of these enzymes is that at least one product is released from the enzyme before all of the substrates have bound. This might seem slightly unlikely at a first glance, but it's actually easily explained and quite a common mechanism. Some very familiar enzymes, for instance the serine proteases (trypsin, chymotrypsin, etc.) and the amino transferases work in this way. The process starts by binding of the enzyme to the first substrate in the usual way: E + A (EA) Notice that I've used parentheses around the EA complex to indicate that it is a central complex. The active site is full as this substrate will be converted to product before the second substrate can bind. The active site has no room for the second substrate so it is full. The next reaction is the key to the whole process: (EA) (FP) In this reaction a part of the substrate has been removed from substrate A, converting it to product P. The removed section has become covalently bound to the enzyme to create a new form of the enzyme, enzyme F. The first product of the reaction is now released and the second substrate binds: (FP) F + P F + B (FB) Now the stored section of the first substrate is transferred to the second
substrate to create the second product, which is then released: (FB) (EQ) (EQ) E + Q The animation should help to clarify this. The Cleland plot for a ping-pong enzyme is quite distinctive: as the first upward arrow (product P) is to the left of the first down arrow (substrate B). Now that we're familiar with the principal types of kinetic mechanism we need to think about experimental techniques to distinguish between them. To start with we'll examine the effects of changes in substrate concentration on multisubstrate reactions. Effects of Substrate Concentration in Multisubstrate Systems ________________________________________ With a multisubstrate enzyme, let's say a typical bi bi enzyme: A + B P + Q we can carry out exactly the same experiment. We simply need to keep one substrate (the fixed substrate) at a constant concentration in all our assays, while we vary the concentration of the other substrate (the variable substrate). The results would be exactly the same as you'd expect in a single substrate system and you could use any of the methods that we've studied to calculate the kinetic parameters. What would happen though if you repeated the experiment with an increased concentration of the fixed substrate. Since you're increasing the concentration of a substrate you would expect the velocity to rise and in fact the reaction would be faster at any given concentration of the variable substrate than it was in the previous experiment. So you'd end up with a second set of data which you could use in your chosen plot. The kinetic parameters would change to reflect the change in velocity. If you repeated this at a variety of concentrations of the fixed substrate you would get a series of lines. A typical Lineweaver-Burk plot obtained as a result of this type of experiment can be seen below. Substrate A was used as the variable substrate and substrate B as the fixed substrate. The actual pattern of lines obtained will vary according to the way in which the enzyme interacts with the two substrates and, as we'll see in the next couple of pages, enables us to distinguish between sequential and ping-pong enzymes. In discussing graphs of this type we'll be considering changes in the Vmax and slope of the line. A change in Vmax indicates the effect that a change in the concentration of the fixed substrate on the reaction speed at very high concentrations of the variable substrate. Remember that Vmax is the velocity of the reaction under those conditions. A change in slope indicates the effect of a change in concentration of the fixed substrate on the speed at very low concentrations of the variable substrate. Remember in our discussion of enzyme inhibition we found that the slope was the rate constant at low substrate concentrations. We'll start by considering the expected results of this experiment when carried out with a sequential enzyme. Substrate concentration assays with sequential enzymes ________________________________________ Consider a bi bi ordered sequential reaction: A + B P + Q The Cleland plot for such a reaction would be: We'll discuss the results of a set of enzyme assays in as the variable substrate and substrate B as the fixed At very low A concentrations the rate limiting step of binding of A to the enzyme as the substrate is in very which substrate A is used substrate. the reaction would be the short supply. An increase
in the concentration of B would reduce the concentration of the EA complex in the reaction mixture by binding to it to form EAB complex. Reducing the concentration of the product of the E + A EA reaction will pull it to the right by the Law of Mass Action. So the increase in B has increased the speed of the rate limiting step, and therefore of the overall reaction. As we're looking at effects at low A concentrations this would be seen as a change in the slope of the Lineweaver-Burk plot. At very high A concentrations the rate limiting step would be the EAB EPQ, which is the inherently slowest reaction, or EA + B EAB at low B levels. An increase in B would increase the speed of either of these as a reactant in the second reaction and by the knock on effect of generating more EAB for the central reacton to occur. This would be seen as a change in Vmax as we are considering effects at high A concentration. In summary then, for an ordered sequential reaction, a change in the concentration of the fixed substrate would bring about a change in both the slope and intercept of the Lineweaver-Burk plot and the graph would be similar to that suggested on the previous page. A similar result would be obtained if the assay was reversed and B used as the variable substrate or if the enzyme used a random sequential mechanism. You might like to demonstrate that yourself as an exercise.
Chapter-9 ISOLATION AND PURIFICATION OF ENZYME INTRODUCTION ENZYME is isolated and purified to obtain maximum desired product because they are not utilized during the product formation. And also isolation of enzyme gives the maximum understanding of the behavior of the enzyme in complex system, its regulation mechanism. PURIFIED enzyme is needed for laboratory work and less purified enzyme is needed for commercial purpose. Purifying enzyme is much labor intensive process and main aim is to separate other protein that are not the part of the enzyme. Often during purification enzyme yield is decreases many folds. Purification of enzyme is aimed to get maximum turn over number that is to increase the activity of enzyme in other world to increase its efficiency of conversion of per mole substrate into the product. It helps in increasing the sensitivity of the diagnosis test in the clinics. Other need of the enzyme purification is the study of kinetics that is rate of enzyme and its specificity towards the substrate. Inhibition kinetics helps in functional study of the enzyme. During the disturbance in the metabolic pathway some enzyme may not form and it results in the formation of unwanted product like in the phenylketonuria disease homigenistic acid secretion in the urine results. Therefore for the diagnostic purpose much pure enzyme is needed. FIGURE 8. 1 strategies for enzyme purification. source of enzyme Method of enzyme isolation (cell lysis by osmotic, enzyme, sonication, or homogenization method) Method of separation. [i] Based on size and mass ( centrifugation, GPC gel permeation chromatography,
Dialysis and ultracentrifugation). [ii] Based on polarity (Ion exchange, electrophoresis, isoelectric focusing, hydrophobic interaction chromatography). [iii] Based on changes in solubility ( by change of pH, change in ionic strength (Salting in or salting out). [iv] Based on change in dielectric strength by adding organic solvent. [v] Based on specific binding site. Affinity chromatography. Affinity elution. Dye ligand chromatography. Immuno-adsorption chromatography. Covalent chromatography. (4). Test of purification a). test of purity b). Test of catalytic activity c). Active site titration Test of purity is done by following method 1. Ultrapurification (for impurities < 5%) 2. Electrophoresis (Examining enzyme composed of non identical subunit) 3. SDS-PAGE ( good method for detecting impurities and for detecting damage of non-identical subunit.) 4. Capillary Electrophoresis 5. Isoelectric focusing ( very sensitive method) 6. Mass spectrometry.( power full method ) Source of enzyme Biologically active enzymes may be extracted from any living organism. A very wide range of sources are used for commercial enzyme production from Actinoplanes to Zymomonas, from spinach to snake venom. Of the hundred or so enzymes being used industrially, over a half are from fungi and yeast and over a third are from bacteria with the remainder divided between animal (8%) and plant (4%) sources (Table 8.1). A very much larger number of enzymes find use in chemical analysis and clinical diagnosis. Non-microbial sources provide a larger proportion of these, at the present time. Microbes are preferred to plants and animals as sources of enzymes because: 1. they are generally cheaper to produce. 2. their enzyme contents are more predictable and controllable, 3. reliable supplies of raw material of constant composition are more easily arranged, and 4. plant and animal tissues contain more potentially harmful materials than microbes, including phenolic compounds (from plants), endogenous enzyme inhibitors and proteases. 5. Attempts are being made to overcome some of these difficulties by the use of animal and plant cell culture. ________________________________________ Table 2.1. Some important industrial enzymes and their sources. Enzyme a EC number b Source Intra/extra -cellular c Scale of production d Industrial use Animal enzymes Catalase 1.11.1.6 Liver I Chymotrypsin 3.4.21.1 Pancreas Lipasee 3.1.1.3 Pancreas E Food Rennetf
Food E
Leather
3.4.23.4 Abomasum E + Cheese Trypsin 3.4.21.4 Pancreas E Leather Plant enzymes Actinidin 3.4.22.14 Kiwi fruit E Food Amylase 3.2.1.1 Malted barley E +++ Brewing -Amylase 3.2.1.2 Malted barley E +++ Brewing Bromelain 3.4.22.4 Pineapple latex E Brewing Glucanaseg 3.2.1.6 Malted barley E ++ Brewing Ficin 3.4.22.3 Fig latex E Food Lipoxygenase 1.13.11.12 Soybeans I Food Papain 3.4.22.2 Pawpaw latex E ++ Meat Bacterial enzymes Amylase 3.2.1.1 Bacillus E +++ Starch -Amylase 3.2.1.2 Bacillus E + Starch Asparaginase 3.5.1.1 Escherichia coli I Health Glucose isomeraseh 5.3.1.5 Bacillus I ++ Fructose syrup Penicillin amidase 3.5.1.11 Bacillus I Pharmaceutical Proteasei 3.4.21.14 Bacillus E +++ Detergent Pullulanasej 3.2.1.41 Klebsiella E Starch Fungal enzymes Amylase 3.2.1.1 Aspergillus E ++ Baking Aminoacylase 3.5.1.14 Aspergillus I Pharmaceutical Glucoamylasek 3.2.1.3 Aspergillus E +++ Starch Catalase 1.11.1.6 Aspergillus I Food Cellulase 3.2.1.4 Trichoderma E Waste Dextranase 3.2.1.11 Penicillium E Food Glucose oxidase 1.1.3.4 Aspergillus I Food Lactasel 3.2.1.23 Aspergillus E Dairy Lipasee 3.1.1.3 Rhizopus E Food Rennetm 3.4.23.6 Mucor miehei E ++ Cheese Pectinasen 3.2.1.15 Aspergillus E ++ Drinks Pectin lyase 4.2.2.10 Aspergillus E Drinks Proteasem 3.4.23.6 Aspergillus E + Baking Raffinaseo 3.2.1.22 Mortierella I Food Yeast enzymes Invertasep 3.2.1.26 Saccharomyces I/E Confectionery Lactasel 3.2.1.23 Kluyveromyces I/E Dairy Lipasee 3.1.1.3 Candida E Food Raffinaseo 3.2.1.22 Saccharomyces I Food a The names in common usage are given. As most industrial enzymes consist of mixtures of enzymes, these names may vary from the recommended names of their principal component. Where appropriate, the recommended names of this principal
component is given below. b The EC number of the principal component. c I - intracellular enzyme; E - extracellular enzyme. d +++ > 100 ton year-1; ++ > 10 ton year-1; + > 1 ton year-1; - < 1 ton year-1. e triacylglycerol lipase; f chymosin; g Endo-1,3(4)- -glucanase; h xylose isomerase; i subtilisin; j dextrin endo-1,6- -glucosidase; k glucan 1,4- -glucosidase; l -galactosidase; m microbial aspartic proteinase; n polygalacturonase; o -galactosidase; p -fructofuranosidase. In practice, the great majority of microbial enzymes come from a very limited number of genera, of which Aspergillus species, Bacillus species and Kluyveromyces (also called Saccharomyces) species predominate. Most of the strains used have either been employed by the food industry for many years or have been derived from such strains by mutation and selection. There are very few examples of the industrial use of enzymes having been developed for one task. Shining examples of such developments are the production of high fructose syrup using glucose isomerase and the use of pullulanase in starch hydrolysis. Media for enzyme production Detailed description of the development and use of fermentors for the large-scale cultivation of microorganisms for enzyme production is outside the scope of this volume but mention of media use is appropriate because this has a bearing on the cost of the enzyme and because media components often find their way into commercial enzyme preparations. Details of components used in industrial scale fermentation broths for enzyme production are not readily obtained. This is not unexpected as manufacturers have no wish to reveal information that may be of technical or commercial value to their competitors. Also some components of media may be changed from batch to batch as availability and cost of, for instance, carbohydrate feedstock change. Such changes reveal themselves in often quite profound differences in appearance from batch to batch of a single enzyme from a single producer. The effects of changing feedstock must be considered in relation to downstream processing. If such variability is likely to significantly reduce the efficiency of the standard methodology, it may be economical to use a more expensive defined medium of easily reproducible composition. Clearly defined media are usually out of the question for large scale use on cost grounds but may be perfectly acceptable when enzymes are to be produced for high value uses, such as analysis or medical therapy where very pure preparations are essential. Less-defined complex media are composed of ingredients selected on the basis of cost and availability as well as composition. Waste materials and byproducts from the food and agricultural industries are often major ingredients. Thus molasses, corn steep liquor, distillers soluble and wheat bran is important components of fermentation media providing carbohydrate, minerals, nitrogen and some vitamins. Extra carbohydrate is usually supplied as starch, sometimes refined but often simply as ground cereal grains. Soybean meal and ammonium salts are frequently used sources of additional nitrogen. Most of these materials will vary in quality and composition from batch to batch causing changes in enzyme productivity. Often the enzyme may be purified several hundred-fold but the yield of the enzyme may be very poor, frequently below 10% of the activity of the original material (Table 2.2). In contrast, industrial enzymes will be purified as little as possible, only other enzymes and material likely to interfere with the process
which the enzyme is to catalyze, will be removed. Unnecessary purification will be avoided as each additional stage is costly in terms of equipment, manpower and loss of enzyme activity. As a result, some commercial enzyme preparations consist essentially of concentrated fermentation broth, plus additives to stabilize the enzyme's activity. The content of the required enzyme should be as high as possible (e.g. 10% w/w of the protein) in order to ease the downstream processing task. This may be achieved by developing the fermentation conditions or, often more dramatically, by genetic engineering. It may well be economically viable to spend some time cloning extra copies of the required gene together with a powerful promoter back into the producing organism in order to get 'over-producers' (see Chapter protein engineering). It is important that the maximum activity is retained during the preparation of enzymes. Enzyme inactivation can be caused by heat, proteolysis, sub-optimal pH, oxidation, denaturants, irreversible inhibitors and loss of cofactors or coenzymes. Of this heat inactivation, this together with associated pH effects is probably the most significant. It is likely to occur during enzyme extraction and purification if insufficient cooling is available, but the problem is less when preparing thermophilic enzymes. Proteolysis is most likely to occur in the early stages of extraction and purification when the proteases responsible for protein turnover in living cells are still present. It is also the major reason for enzyme inactivation by microbial contamination. In their native conformations, enzymes have highly structured domains which are resistant to attack by proteases because many of the peptide bonds are mechanically inaccessible and because many proteases are highly specific. The chances of a susceptible peptide bond in a structured domain being available for protease attack are low. Single 'nicks' by proteases in these circumstances may have little immediate effect on protein conformation and, therefore, activity. The effect, however, may severely reduce the conformational stability of the enzyme to heat or pH variation so greatly reducing its operational stability. If the domain is unfolded under these changed conditions, the whole polypeptide chain may be available for proteolysis and the same, specific, protease may destroy it. Clearly the best way of preventing proteolysis is to rapidly remove, or inhibit, protease activity. Before this can be achieved it is important to keep enzyme preparations cold to maintain their native conformation and slow any protease action that may occur. Some intracellular enzymes are used commercially without isolation and purification but the majority of commercial enzymes is either produced extracellularly by the microbe or plant or must be released from the cells into solution and further processed (Figure 2.1). Solid/liquid separation is generally required for the initial separation of cell mass, the removal of cell debris after cell breakage and the collection of precipitates. This can be achieved by filtration, centrifugation or aqueous biphasic partition. In general, filtration or aqueous biphasic systems are used to remove unwanted cells or cell debris whereas centrifugation is the preferred method for the collection of required solid material. Figure 2.1. Flow diagram for the preparation of enzymes. The main objective of the purification is To obtained maximum possible yield. It should have maximum possible purity. It should have maximum possible activity. Cell can be break down byfollowing methods 1. Osmotic shock 2. Enzymic lysis. 3. Homogenization 4. Ultracentrifugation. A. Cell breakage by osmotic shock Various intracellular enzymes are used in significant quantities and must be
released from cells and purified (Table 2.1). The amount of energy that must be put into the breakage of cells depends much on the type of organism and to some extent on the physiology of the organism. Some types of cell are broken readily by gentle treatment such as osmotic shock (e.g. animal cells and some gram-negative bacteria such as Azotobacter species), whilst others are highly resistant to breakage. These include yeasts, green algae, fungal mycelia and some gram-positive bacteria which have cell wall and membrane structures capable of resisting internal osmotic pressures of around 20 atmospheres (2 MPa) and therefore have the strength, weight for weight, of reinforced concrete. Consequently a variety of cell disruption techniques have been developed involving solid or liquid shear or cell lysis. The rate of protein released by mechanical cell disruption is usually found to be proportional to the amount of releasable protein. (2.5) where P represents the protein content remaining associated with the cells, t is the time and k is a release constant dependent on the system. Integrating from P = Pm (maximum possible protein releasable) at time zero to P = Pt at time t gives (2.6) (2.7) As the protein released from the cells (Pr) is given by (2.8) the following equation for cell breakage is obtained (2.9) It is most important in choosing cell disruption strategies to avoid damaging the enzymes. The particular hazards to enzyme activity relevant to cell breakage are summarized in Table 2.3. The most significant of these, in general, are heating and shear. ________________________________________ Table 2.3. Hazards likely to damage enzymes during cell disruption. Heat All mechanical methods require a large input of energy, generating heat. Cooling is essential for most enzymes. The presence of substrates, substrate analogues or polyols may also help stabilise the enzyme. Shear Shear forces are needed to disrupt cells and may damage enzymes, particularly in the presence of heavy metal ions and/or an air interface. Proteases Disruption of cells will release degradative enzymes which may cause serious loss of enzyme activity. Such action may be minimised by increased speed of processing with as much cooling as possible. This may be improved by the presence of an excess of alternative substrates (e.g. inexpensive protein) or inhibitors in the extraction medium. pH Buffered solutions may be necessary. The presence of substrates, substrate analogues or polyols may also help stabilise the enzyme. Chemical Some enzymes may suffer conformational changes in the presence of detergent and/or solvents. Polyphenolics derived from plants are potent inhibitors of enzymes. This problem may be overcome by the use of adsorbents, such as polyvinylpyrrolidone, and by the use of ascorbic acid to reduce polyphenol oxidase action. Oxidation Reducing agents (e.g. ascorbic acid, mercaptoethanol and dithiothreitol) may be necessary. Foaming The gas-liquid phase interfaces present in foams may disrupt enzyme conformation. Heavy-metal toxicity Heavy metal ions (e.g. iron, copper and nickel) may be introduced by leaching from the homogenisation apparatus. Enzymes may be protected from irreversible inactivation by the use of chelating reagents, such as EDTA. ________________________________________ Media for enzyme extraction will be selected on the basis of cost-effectiveness so will include as few components as possible. Media will usually be buffered at a pH value which has been determined to give the maximum stability of the enzyme to be extracted. Other components will combat other hazards to the enzyme, primarily factors causing denaturation (Table 2.3).
B. Use of enzymic lytic methods The breakage of cells using non-mechanical methods is attractive in that it offers the prospects of releasing enzymes under conditions that are gentle, do not subject the enzyme to heat or shear, may be very cheap, and are quiet to the user. The methods that are available include osmotic shock, freezing followed by thawing, cold shock, desiccation, enzymic lysis and chemical lysis. Each method has its drawbacks but may be particularly useful under certain specific circumstances. Certain types of cell can be caused to lyse by osmotic shock. This would be a cheap, gentle and convenient method of releasing enzymes but has not apparently been used on a large scale. Some types of cell may be caused to autolyse, in particular yeasts and Bacillus species. Yeast invertase preparations employed in the industrial manufacture of invert sugars are produced in this manner. Autolysis is a slow process compared with mechanical methods, and microbial contamination is a potential hazard, but it can be used on a very large scale if necessary. Where applicable, desiccation may be very useful in the preparation of enzymes on a large scale. The rate of drying is very important in these cases, slow methods being preferred to rapid ones like lyophilisation. Enzymic lysis using added enzymes has been used widely on the laboratory scale but is less popular for industrial purposes. Lysozyme, from hen egg-white, is the only lytic enzyme available on a commercial scale. It has often used to lyse Gram positive bacteria in an hour at about 50,000 U Kg-1 (dry weight). The chief objection to its use on a large scale is its cost. Where costs are reduced by the use of the relatively inexpensive, Lysozyme-rich, dried egg white, a major separation problem may be introduced. Yeast-lytic enzymes from Cytophaga species have been studied in some detail and other lytic enzymes are under development. If significant markets for lytic enzymes are identified, the scale of their production will increase and their cost is likely to decrease.Lysis by acid, alkali, surfactants and solvents can be effective in releasing enzymes, provided that the enzymes are sufficiently robust. Detergents, such as Triton X-100, used alone or in combination with certain chaotropic agents, such as guanidine HCl, are effective in releasing membrane-bound enzymes. However, such materials are costly and may be difficult to remove from the final product. c. Ultrasonic cell disruption The treatment of microbial cells in suspension with inaudible ultrasound (greater than about 12 kHz) results in their inactivation and disruption. Ultrasonication utilises the rapid sinusoidal movement of a probe within the liquid. It is characterised by high frequency (12 kHz - 1 MHz), small displacements (less than about 50 m), moderate velocities (a few m s-1), steep transverse velocity gradients (up to 4,000 s-1) and very high acceleration (up to about 20,000 g). Ultrasonication produces cavitation phenomena when acoustic power inputs are sufficiently high to allow the multiple production of microbubbles at nucleation sites in the fluid. The bubbles grow during the rarefying phase of the sound wave, then are collapsed during the compression phase. On collapse, a violent shock wave passes through the medium. The whole process of gas bubble nucleation, growth and collapse due to the action of intense sound waves is called cavitation. The collapse of the bubbles converts sonic energy into mechanical energy in the form of shock waves equivalent to several thousand atmospheres (300 MPa) pressure. This energy imparts motions to parts of cells which disintegrate when their kinetic energy content exceeds the wall strength. An additional factor which increases cell breakage is the microstreaming (very high velocity gradients causing shear stress) which occur near radially vibrating bubbles of gas caused by the ultrasound. Much of the energy absorbed by cell suspensions is converted to heat so effective cooling is essential. The amount of protein released by sonication has been shown to follow Equation 2.9. The constant (k) is independent of cell concentrations up to high levels and approximately proportional to the input acoustic power above the threshold power necessary for cavitation. Disintegration is independent of the
sonication frequency except insofar as the cavitation threshold frequency depends on the frequency. Equipment for the large-scale continuous use of ultrasonic has been available for many years and is widely used by the chemical industry but has not yet found extensive use in enzyme production. Reasons for this may be the conformational lability of some (perhaps most) enzymes to sonication and the damage that they may realize though oxidation by the free radicals, singlet oxygen and hydrogen peroxide that may be concomitantly produced. Use of radical scavengers (e.g. N2O) have been shown to reduce this inactivation. As with most cell breakage methods, very fine cell debris particles may be produced which can hinder further processing. Sonication remains, however, a popular, useful and simple small-scale method for cell disruption. d. Cell lysis by High pressure homogenisers Various types of high pressure homogeniser are available for use in the food and chemicals industries but the design which has been very extensively used for cell disruption is the Manton-Gaulin APV type homogeniser. This consists of a positive displacement pump which draws cell suspension (about 12% w/v) through a check valve into the pump cylinder and forces it, at high pressures of up to 150 MPa (10 tons per square inch) and flow rates of up to 10,000 L hr-1, through an adjustable discharge valve which has a restricted orifice (Figure 2.5). Cells are subjected to impact, shear and a severe pressure drop across the valve but the precise mechanism of cell disruption is not clear. The main disruptive factor is the pressure applied and consequent pressure drop across the valve. This causes the impact and shear stress which are proportional to the operating pressure. Figure 2.2. A cross-section through the Manton-Gaulin homogeniser valve, showing the flow of material. The cell suspension is pumped at high pressure through the valve impinging on it and the impact ring. The shape of the exit nozzle from the valve seat varies between models and appears to be a critical determinant of the homogenisation efficiency. The model depicted is the 'CD Valve' from APV Gaulin. ________________________________________ As narrow orifices which are vulnerable to blockage are key parts of this type of homogeniser, it is unsuitable for the disruption of mycelial organisms but has been used extensively for the disruption of unicellular organisms. The release of proteins can be described by Equation 2.9 but normally a similar relationship is used where the time variable is replaced by the number of passes (N) through the homogeniser. (2.10) In the commonly-used operating range with pressures below about 75 MPa, the release constant (k) has been found to be proportional to the pressure raised to an exponent dependent on the organism and its growth history (e.g. k=k'P2.9 in Saccharomyces cerevesiae and k=k'P2.2 in Escherichia coli, where P represents the operating pressure and k' is a rate constant). Different growth media may be selected to give rise to cells of different cell wall strength. Clearly, the higher the operating pressure, the more efficient is the disruption process. The protein release rate constant (k) is temperature dependent, disruption being more rapid at higher temperatures. In practice, this advantage cannot be used since the temperature rise due to adiabatic compression is very significant so samples must be pre-cooled and cooled again between multiple passes. At an operating pressure of 50 MPa, the temperature rise each pass is about 12 deg. C. In addition to the fragility of the cells, the location of an enzyme within the cells can influence the conditions of use of an homogeniser. Unbound intracellular enzymes may be released by a single pass whereas membrane bound enzymes require several passes for reasonable yields to be obtained. Multiple passes are undesirable because, of course, they decrease the throughput productivity rate and because the further passage of already broken cells results in fine debris which is excessively difficult to remove further downstream. Consequently, homogenisers
will be used at the highest pressures compatible with the reliability and safety of the equipment and the temperature stability of the enzyme(s) released. High pressure homogenisers are acceptably good for the disruption of unicellular organisms provided the enzymes needed are not heat labile. The shear forces produced are not capable of damaging enzymes free in solution. The valve unit is prone to erosion and must be precision made and well maintained. e. Use of bead mills for cell lysis When cell suspensions are agitated in the presence of small steel or glass beads (usually 0.2 -.1.0 mm diameter) they are broken by the high liquid shear gradients and collision with the beads. The rate and effectiveness of enzyme release can be modified by changing the rates of agitation and the size of the beads, as well as the dimensions of the equipment. Any type of biomass, filamentous or unicellular, may be disrupted by bead milling but, in general, the larger sized cells will be broken more readily than small bacteria. For the same volume of beads, a large number of small beads will be more effective than a relatively small number of larger beads because of the increased likelihood of collisions between beads and cells. Bead mills are available in various sizes and configurations from the Mickle shaker which has a maximum volume of about 40 ml to continuous process equipment capable of handling up to 200 Kg wet yeast or 20 Kg wet bacteria each hour. The bead mills that have been studied in most detail are the Dyno-Mill and the NetschMolinex agitator, both of which consist of a cylindrical vessel containing a motor-driven central shaft equipped with impellers of different types. Both can be operated continuously, being equipped with devices which retain the beads within the milling chamber. Glass Ballotini or stainless steel balls are used, the size range being selected for most effective release of the enzyme required. Thus 1 mm diameter beads are satisfactory for the rapid release of periplasmic enzymes from yeast but 0.25 mm diameter beads must be used, for a longer period, to release membrane-bound enzymes from bacteria. The kinetics of protein release from bead mills follows the relationship given by Equation 2.9 with respect to the time (t) that a particle spends in the mill. Unfortunately, however well designed these mills are, when continuously operated there will be a significant amount of backmixing which reduces the efficiency of the protein released with respect to the average residence time () . This is more noticeable at low flow rates (high average residence times) and when the proportion of protein released is high. It may be counteracted by designing the bead mill to encourage plug flow characteristics. Under these circumstances the relationship can be shown to be (2.11) where i represents the degree of backmixing (i.e. i = 0 under ideal plug flow conditions and i = 1 for ideal complete backmixing). Equation 2.11 reduces to give the simplified relationship of Equation 2.9 at low (near zero) values of i. In addition to bead size, the protein release rate constant (k) is a function of temperature, bead loading, impeller rotational speed and cell loading. Impeller speeds can be increased with advantage until bead breakage becomes significant but heat generation will also increase. At a constant impeller speed, the efficiency of the equipment declines with throughput as the degree of backmixing increases. There will be an optimum impeller tip speed at which the increases in disruption are balanced by increases in backmixing. In general, increased bead loading increases the rate of protein release but also increases the production of heat and the power consumption. Heat production is the major problem in the use of bead mills for enzyme release, particularly on a large (e.g. 20 liters) scale. Smaller vessels may be cooled adequately through cooling jackets around the bead chamber but larger mills require cooling through the agitator shaft and impellers. However, if cooling is effective there is little damage to the enzymes released. 2.6 Method of separation of enzyme from lysed cell; Centrifugation
B. ULTRACENTRIFUGATION This technique is used to investigate the different parameter of the molecule such as molecular mass, shape, and density. In this process gravity plays most important role. Therefore, the basis of centrifugation separation techniques therefore is to exert the lager force than does the gravitational earth, thus increasing the rate at which the particle settles. The technique can be divided into two types: 1 preparative centrifugation: for separation of the whole cells, subcellular organelles, plasma membranes, polysomes, ribosome, chromatin, nucleic acids, lipoprotein and viruses 2. Analytical centrifugation: it is devoted to study of pure or virtually pure or macromolecular or particles. They are related with the study of macromolecular structure rather than the collection of particle fractions. Principles: Rate depends on the centrifugal field G directed radially outward (angular velocity) G = r 2 =2/60 revolution min-1 ultracentrifuge have maximum speed of 600,000 g. the chamber is surrounded by the refrigerated, sealed and evacuated to minimized excess rise in temperature. Note that there must be equal loading of sample. For safety purpose ultracentrifuge are always enclosed in heavy armour plating. This is type of preparative ultracentrifuge, better for the deproteinisation of physiological fluids for amino acid analysis. Another type of ultracentrifuge is analytical type. It has speed 70,000 rev per min (500,000 g). its fabrication is of same type. It can also measure the refractive index, and is equipped with the ultraviolet light absorption system. Light of suitable wavelength is passed through moving analytical cell containing the solution under analysis e.g. protein or nucleic acid and intensity of light is measured on the photographic plate. Optical system records the change in refractive index. The Schleiren optical system plots the refractive index gradient against distance along the analytical cell, which makes it useful in analysis in the locating in boundaries in sedimentation velocity measurements. A. Differential centrifugation This method is based upon the difference in the sedimentation rate of particles of different size and density. In centrifugation the larger particle are sedimented first. The particle having the same mass but different density, the denser particle is sedimented first and less dense will sediment later. Particle having similar density can be separated by the differential centrifugation or the rate zonal method. In differentiated centrifugation, the particle to be separated is divided centrifugally into a number of fractions by increasing the applied centrifugal field. After the centrifugation, pellet and supernatant are separated, pellet is washed several time and again centrifugation is done. The particle moves against respective sedimentation rates. Centrifugation is continued long enough to pellet all the largest class of particles, the resulting supernatant then being centrifuged at higher speed to separate medium sized particle and so on. However, since particles of varying sizes and densities were distributed homogeneously at the start of centrifugation, so pellet will not be homogeneous but will contain a mixture of all the sediment components. Particle separation by the rate zonal technique is based on differences in the size, shape, and density of the particle. Most of the biological particles having same size are having narrower density range. So , separation of similar particle by the rate zonal technique is based mainly upon differences in their sizes and
can not be separated easily like mitochondria, lysosome, peroxisomes. The technique has been used for the separation of enzymes hormones and RNA and DNA hybrids, ribosomal subunits, sub cellular organelles. There are two type of density gradient centrifugation, --1. The rate zonal technique and the isopycnic (equal density) Isopycnic centrifugation depends solely upon the buoyant density of the particles and not its shape or size and is independent of time the size of the particle affecting only the rate at which it reaches its isopycnic position in the gradient. The technique is useful in separating the particle of same size but differing in density. 1Svedberg = 10-13 second Centrifugation separates on the basis of the particle size and density difference between the liquid and solid phases. Sedimentation of material in a centrifugal field may be described by (2.11) where v is the rate of sedimentation, d is the particle diameter, rs is the particle density, rl is the solution density is the angular velocity in radians s1, r is the radius of rotation, is the kinematic viscosity, Fs is a correction factor for particle interaction during hindered settling and is a shape factor (=1 for spherical particles). Fs depends on the volume fraction of the solids present; approximately equaling 1, 0.5, 0.1 and 0.05 for 1%, 3%, 12% and 20% solids volume fraction respectively. Only material which reaches a surface during the flow through continuous centrifuges will be removed from the centrifuge feedstock, the efficiency depending on the residence time within the centrifuge and the distance necessary for sedimentation (D). This residence time will equal the volumetric throughput () divide by the volume of the centrifuge (V). The maximum throughput of a centrifuge for efficient use is given by (2.12) The efficiency of the process is seen to depend on the solids volume fraction, the effective clarifying surface (V/D) and the acceleration factor (2r/g, where g is the gravitational constant, 921 cm s-2; a rotor of radius 25 cm spinning at 1 rev s-1 has an acceleration factor of approximately 1 G). Low acceleration factors of about 1 500 g may be used for harvesting cells whereas much higher acceleration factors are needed to collect enzyme efficiently. The product of these factors (2rV/gD) is called the sigma factor and is used to compare centrifuges and to assist scale-up. Laboratory centrifuges using tubes in swing-out or angle head rotors have high angular velocity and radius of rotation (r) but small capacity (V) and substantial sedimentation distance (D). This type of design cannot be scaled-up safely, primarily because the mechanical stress on the centrifuge head increases with the square of the radius, which must increase with increasing capacity. For large-scale use, continuous centrifuges of various types are employed (Figure 2.2). These allow the continuous addition of feedstock, the continuous removal of supernatant and the discontinuous, semicontinuous or continuous removal of solids. Where discontinuous or semicontinuous removal of precipitate occurs, the precipitate is flushed out by automatic discharge systems which cause its dilution with water or medium and may be a problem if the precipitate is required for further treatment. Centrifugation is the generally preferred method for the collection of enzyme-containing solids as it does not present a great hazard to most enzymes so long as foam production, with consequent enzymic inactivation, is minimised. centrifuges are long and thin enabling rapid acceleration and deceleration, minimizing the down-time required for the removal of the sedimented solids. Here the radius and effective liquid thickness are both small allowing a high angular velocity and hence high centrifugal force; small models can be used at acceleration factors up to 50,000 g, accumulating 0.1 Kg of wet deposit whereas
large models, designed to accumulate up to 5 Kg of deposit, are restricted to 16,000 g. The capacities of these centrifuges are only moderate.Multichamber discstack centrifuges, originally designed (by Westfalia and Alpha-Laval) for cream separation, contain multiple coned discs in a stack which are spun and on which the precipitate collects. They may be operated either semi-continuously or, by using a centripetal pressurising pump within the centrifuge bowl which forces the sludge out through a valve, continuously. The capacity and radius of such devices are large and the thickness of liquid is very small, due to the large effective surface area. The angular velocity, however, is restricted giving a maximum acceleration factor of about 2,000 g. A different design which is rather similar in principle is the solid bowl scroll centrifuge in which an Archimedes' screw collects the precipitate so that fluid and solids leave at opposite ends of the apparatus. These can only be used at low acceleration (about 3,000 g) so they are suitable only for the collection of comparatively large particles. Although many types of centrifuge are available, the efficient precipitation of small particles of cell debris can be difficult, sometimes near-impossible. Clearly from Equation 2.2, the efficiency of centrifugation can be improved if the particle diameter (d) is increased. This can be done either by coagulating or flocculating particles. Coagulation is caused by the removal of electrostatic charges (e.g. by pH change) and allowing particles to adhere to each other. Flocculation is achieved by adding small amounts of high-molecular-weight charged materials which bridge oppositely-charged particles to produce a loose aggregate which may be readily removed by centrifugation or filtration. Flocculation and coagulation are cheap and effective aids to precipitating or otherwise harvesting whole cells, cell debris or soluble proteins but, of course, it is essential that the agents used must not inhibit the target enzymes. It is important to note that the choice of flocculants is determined by the pH and ionic strength of the solution and the nature of the particles. Most flocculants have very definite optimum concentrations above which further addition may be counter-effective. Some flocculants can be rapidly ruined by shear. A comparatively recent introduction designed for the removal of cell debris is a moderately hydrophobic product in which cellulose is lightly derivatised with diethylaminoethyl functional groups. This material (Whatman CDR; cell debris remover) is inexpensive (essential as it is not reusable), binds to unwanted negatively charged cell constituents, acts as a filter aid and may be incinerated to dispose of hazardous 2.7 Filtration of enzyme after separation Filtration separates simply on the basis of particle size. Its efficiency is limited by the shape and compressibility of the particles, the viscosity of the liquid phase and the maximum allowable pressures. Large-scale simple filtration employs filter cloths and filter aids in a plate and frame press configuration, in rotary vacuum filters or centrifugal filters (Figure 2.3). The volumetric throughput of a filter is proportional to the pressure (P) and filter area (AF) and inversely proportional to the filter cake thickness (DF) and the dynamic viscosity (2.13) Where, k is a proportionality constant dependent on the size and nature of the particles. For very small particles k depends on the fourth power of their diameter. Filtration of particles that are easily compressed leads to filter blockage and the failure of Equation 2.3 to describe the system. Under these circumstances a filter aid, such as celite, is mixed with the feedstock to improve the mechanical stability of the filter cake. Filter aids are generally used only where the liquid phase is required as they cause substantial problems in the recovery of solids. They also may cause loss of enzyme activity from the solution due to physical hold-up in the filter cake. It is often difficult for a process development manager to decide whether to attempt to recover enzyme trapped in this way. Problems associated with the build-up of the filter cake may also be avoided
by high tangential flow of the feedstock across the surface of the filter, a process known as crossflow microfiltration (Figure 2.4). This method dispenses with filter aids and uses special symmetric microporous membrane assemblies capable of retaining particles down to 0.1 - 1 m diameter (cf. Bacillus diameter of about 2 m). ________________________________________ Figure 2.3. The basic design of the rotary vacuum filter. The suspension is sucked through a filter cloth on a rotating drum. This produces a filter cake which is removed with a blade. The filter cake may be rinsed during its rotation. These filters are generally rather messy and difficult to contain making them generally unsuitable for use in the production of toxic or recombinant DNA products. There have been recent developments that improve their suitability, however, such as the Disposable Rotary Drum Filter. ________________________________________ A simple and familiar filtration apparatus is the perforate bowl centrifuge or basket centrifuge, in effect a spin drier. Cell debris is collected on a cloth with, or without, filter aid and can be skimmed off when necessary using a suitable blade. Such centrifugal filters have a large radius and effective liquid depth, allowing high volumes. However, safety decrees that the angular velocity must be low and so only large particles (e.g. plant material) can be removed satisfactorily. ________________________________________ Figure 2.4. Principles of (a) dead-end filtration and (b) cross-flow filtration. In dead-end filtration the flow causes the build-up of the filter cake, which may prevent efficient operation. This is avoided in cross-flow filtration where the flow sweeps the membrane surface clean. ________________________________________ Chapter-10 Separation in Aqueous biphasic systems The 'incompatibility' of certain polymers in aqueous solution was first noted by Beijerinck in 1296. In this case two phases were formed when agar was mixed with soluble starch or gelatin. Since then, many two phase aqueous systems have been found; the most thoroughly investigated being the aqueous dextran-polyethylene glycol system (e.g. 10% polyethylene glycol 4000/2% dextran T500), where dextran forms the more hydrophilic, denser, lower phase and polyethylene glycol the more hydrophobic, less dense, upper phase. Aqueous three phase systems are also known. Phases form when limiting concentrations of the polymers are exceeded. Both phases contain mainly water and are enriched in one of the polymers. The limiting concentrations depend on the type and molecular weight of the polymers and on the pH, ionic strength and temperature of the solution. Some polymers form the upper hydrophobic phase in the presence of fairly concentrated solutions of phosphates or sulphate (e.g. 10% polyethylene glycol 4000/12.5% potassium phosphate buffer). A drawback to the useful dextran/polyethylene glycol system is the high cost of the purified Dextran used. This has been alleviated by the use of crude unfractionated dextran preparations, much cheaper hydroxypropyl starch derivatives and salt-containing biphasic systems. Aqueous biphasic systems are of considerable value to biotechnology. They provide the opportunity for the rapid separation of biological materials with little probability of denaturation. The interfacial tension between the phases is very low (i.e. about 400-fold less than that between water and an immiscible organic solvent), allowing small droplet size, large interfacial areas, efficient mixing under very gentle stirring and rapid partition. The polymers have a stabilizing influence on most proteins. A great variety of separations have been achieved, by far the most important being the separation of enzymes from broken crude cell material. Separation may be achieved in a few minutes, minimizing the harmful action of endogenous proteases. The systems have also been used successfully for
the separation of different types of cell membranes and organelles, the purification of enzymes and for extractive bioconversions. Continuous liquid twophase separation is easier than continuous solid/liquid separation using equipment familiar from immiscible solvent systems, for example disc-stack centrifuges and counter-current separators. Such systems are readily amenable to scale-up and may be employed in continuous enzyme extraction processes involving some recycling of the phases. Cells, cell debris proteins and other material distribute themselves between the two phases in a manner described by the partition coefficient (P) defined as. --------------------(2.14) Where Ct and Cb represent the concentrations in the top and bottom phases respectively. The yield and efficiency of the separation is determined by the relative amounts of material in the two phases and therefore depends on the volume ratio (Vt/Vb). The partition coefficient is exponentially related to the surface area (and hence molecular weight) and surface charge of the particles in addition to the difference in the electrical potential and hydrophobicity of the phases. It is not generally very sensitive to temperature changes. This means that proteins and larger particles are normally partitioned into one phase whereas smaller molecules are distributed more evenly between phases. A partition coefficient of greater than 3 is required if usable yields are to be achieved by a single extraction process. Typical partition coefficients for proteins are 0.01-100 whereas the partition coefficients for cells and cell debris are effectively zero. The influence of pH and salts on protein partition is complex, particularly when phosphate buffers are present. A given protein distributes differently between the phases at different pH's and ionic strength but the presence of phosphate ions affect the partition coefficient in an anomalous fashion because these ions distribute themselves unequally resulting in electrostatic potential (and pH) differences. This means that systems may be 'tuned' to enrich an enzyme in one phase, ideally the upper phase with cell debris and unwanted enzymes in the lower phase. An enzyme may be extracted from the upper (polyethylene glycol) phase by the addition of salts or further polymer, generating a new biphasic system. This stage may be used to further purify the enzyme. A powerful modification of this technique is to combine phase partitioning and affinity partitioning. Affinity ligand (e.g. triazine dyes) may be coupled to either polymer in an aqueous biphasic system and thus greatly increase the specificity of the extraction. Phases form when limiting concentrations of the polymers are exceeded. Both phases contain mainly water (typically 70-90% w/w water) and are enriched in one of the polymers. The limiting concentrations depend on the type and molecular weight of the polymers and on the pH, ionic strength and temperature of the solution. Some polymers form a two-phase system by themselves; PEG forming the upper morehydrophobic phase in the presence of fairly concentrated solutions of citrates, phosphates or sulfates or at higher temperatures (see below). Such aqueous liquidliquid two-phase systems are finding increasing use in the extractive separation of labile biomolecules such as proteins, offering mild conditions due to the low interfacial tension between the phases (i.e. about 400-fold less than that between water and an immiscible organic solvent) allowing small droplet size, large interfacial areas, efficient mixing under very gentle stirring and rapid partition. The polymers also have a stabilizing influence on most proteins. A great variety of separations have been achieved, by far the most important being the separation of enzymes from broken crude cell material. Separation may be achieved in a few minutes, minimizing the harmful action of endogenous proteases. The systems have also been used successfully for the separation of different types of cell membranes, organelles and actinide ions, the purification of enzymes, extractive bioconversions. Although sometimes perceived as due to polymer incompatibilities, the properties of these biphasic systems can be mainly attributed to incompatibility between aqueous pools of low and higher density water. Each phase may be considered as a different, although aqueous, solvent with
properties determined by its structuring. PEG usually has a far higher concentration in the upper (low-density) phase of such solutions in spite of its inherent density being greater than water. These, together with the properties of this PEG phase encourage the belief that it creates a predominantly low-density water environment due to its partially hydrophobic character, in turn mainly determined by the methylene groups. Further proof of this may be seen by use of microwave dielectric measurements, which show the water surrounding PEG to be ordered, whereas that surrounding more hydrophilic polymers is disordered [332]. Also, the dissolution of PEG is exothermic (and increasingly exothermic with PEG size), in line with a shift in the ES CS equilibrium towards the more ordered ES structure. It is interesting and perhaps not simply fortuitous that the diameter (4.9 ) of the favored PEG helix (formed by trans, gauche, trans links across the C-O-C-C, O-C-C-O, C-C-O-C bonds) is the same as the diameter of the spines of the ES water cluster (4.7 ) formed by pentagonal boxes, the ether (O-C-C-O) distances (2.22 ) are close to the OO distances (2.24 ) in water and the next ether (O-C-C-O-C-C-O) (5.6 ) distances are close to the next vertex distance on opposite sides of the pentagonal boxes (5.4 ).a Model building shows that optimum hydrogen bonding would tend to distort this PEG helix, however. The strongly-held hydration, as determined by viscosity, increases from two molecules of water per PEG monomer at very low polymerization (tetramer) to 5 molecules of water per PEG monomer for 45-mer , showing that the extent of water clustering increases with PEG size. The partitioning of proteins into the hydrophobic PEG phase shows great sensitivity to the protein's surface hydrophobicity (partition increasing with surface hydrophobicity) and also depends on the PEG size; increasing with PEG molecular mass , in line with the extent of water clustering. Increasing PEG size and concentration both increase the proteins' effective hydration as the PEG is excluded from the proteins' surface. However, when the PEG phase becomes too ordered (e.g. at higher PEG size) partitioned proteins are excluded due to the reduced available water content. An interesting and revealing phenomenon occurs in PEG solutions as the temperature is raised; the solution at low temperatures separates into two phases (PEG-rich and PEG-poor) at higher temperature (separating at the cloud-point) and reverts to a single phase at even higher temperatures. This may be explained as the PEG creating a low-density water environment with decreased entropy. At low temperatures a solution is formed due to the enthalpy of hydrogen bonding between the PEG and the water more than compensating for the entropy lost in forming the low-density water. This entropy loss is required, due to the hydrophobicity of the methylene groups, but is not great as the water is somewhat ordered already at lower temperatures. At the cloud point, the entropy cost is greater as the water is no longer naturally as structured, and two phases develop. The stronger hydrogen bonding in D2O, relative to H2O, is expected to raise this cloudpoint. At higher temperatures still, the water possesses excess energy and cannot be structured by the PEG. This reduces the entropic cost, so allowing a solution to form once more. Anions have a distinct effect on the cloud point in line with the Hofmeister Series (cloud point lowering: SCN- < I- < Br- < Cl- < F- < OH- < SO42- < HPO42- < CO32- < PO43-); the greater lowering of the cloud point is in line with greater surface charge density , stronger hydration, greater tendency to avoid low-density water and the greater destruction of the natural structuring of the water. A oppositely-ordered compensating effect on the cloud point has been recognized due to binding of the anions to the polymer surface. This tends to raise the cloudpoint at lower salt concentrations as the bound salt increases the polymer net charge and, hence, solubility. The relative effect of the ions is the reverse of the Hofmeister series just given with weakly hydrated ions binding best, i.e. SCN- having the greatest effect and ionic kosmotropes below Cl- having negligible effect. Cations have a lesser but opposite effect to anions with chaotropes (e.g. NH4+) tending to lower the cloud point but kosmotropes (e.g. Li+) raising it.
Exceptionally, however, some di- and trivalent cations such as Mg2+ and Zn2+ act counter to their normal Hofmeister behavior, due presumably to their specific chelation to oxygen atoms in the PEG molecules. Anions and cations distribute themselves differently between the phases depending on their affinity for low or higher density water but with the requirements that the phases be electrically neutral and iso-osmotic, so producing an interfacial potential difference, which may aid the partitioning of charged biomolecules. Thus sulfate and phosphate ions prefer the bottom phase and, as a consequence, negatively charged proteins are partitioned into the upper PEG phase, so allowing more sulfate or phosphate ions to partition into their preferred lower phase. Preference for the PEG-rich or PEG-poor phase is related to the Hofmeister Series for the structuring ability of the salts, particularly the anions (e.g. preference for PEG-rich phase: I- > Br- > Cl- > F- > SO42-; Cs+ > Na+ > Ba2+ > Ca2+; preference for PEG-poor phase: SO42- > F- > Cl- > Br- > I-). A similar Hofmeister Series effect is noticed intensifying the incompatibility between two polymers such as polyethylenimine-PEG, or dextran-PEG, by increasing the concentration of strongly hydrated (CS-forming) anions, such as sulfate. a Note that the polymers formed with either one (-O-C-O-C-O-) or three (-O-C-C-CO-C-C-C-O-) methylene groups between the oxygen atoms are both insoluble in water. The reason however is not so much that the OO distances (2.12 and 4.79 respectively) fit less well with the water cluster spacing but rather that the molecules form almost-linear extended (rather than helical) chains with a pronounced hydrophobic character that have strong intra-molecular attraction. 2.9 Preparation of enzymes from clarified solution; Ultrafiltration In many cases, especially when extracellular enzymes are being prepared for sale, the clarified solution is simply concentrated, preservative materials added, and sold as a solution or as a dried preparation. The concentration process chosen will be the cheapest which is compatible with the retention of enzyme activity. For some enzymes rotary evaporation can be considered, followed if necessary by spray drying. The most popular method, though, is ultrafiltration, whereby water and low molecular weight materials are removed by passage through a membrane under pressure, enzyme being retained. Ultrafiltration differs from conventional filtration and microfiltration with respect to the size of particles being retained (< 50 nm diameter). It uses asymmetric microporous membranes with a relatively dense but thin skin, containing pores, supported by a coarse strong substructure. Membranes possessing molecular weight cut-offs from 1000 to 100,000 and usable at pressure up to 2 MPa are available. There are number types of apparatus available. Stirred cells represent the simplest configuration of ultrafiltration cell. The membrane rests on a rigid support at the base of a cylindrical vessel which is equipped with a magnetic stirrer to combat concentration polarization. It is not suitable for large scale use but is useful for preliminary studies and for the concentration of laboratory column eluates. Various large-scale units are available in which membranes are formed into wide diameter tubes (1 - 2 cm diameter) and the tubes grouped into cartridges. These are not as compact as capillary systems (area/volume about 25 m1) and are very expensive but are less liable to blockage by stray large particles in the feedstream. Cheaper thin-channel systems are available (area/volume about 500 m-1) which use flat membrane sandwiches in filter press arrangements of various designs chosen to produce laminar flow across the membrane and minimise concentration polarization. Capillary membranes represent a relatively cheap and increasingly popular type of ultrafiltration system which uses micro-tubular membranes 0.2 - 1.1 mm diameter and provides large membrane areas within a small unit volume (area/volume about 1000 m-1). Membranes are usually mounted into modules for convenient manipulation. This configuration of membranes can be scaled up with ease. Commercial models are available that give ultrafiltration rates of up to 600 L hr-1. The steady improvement in the performance, durability and reliability of membranes
has been a boon to enzyme technologists, encouraging wide use of the various ultrafiltration configurations. Problems with membrane blockage and fouling can usually be overcome by treatment of membranes with detergents, proteases or, with care, acids or alkalis. The initial cost of membranes remains considerable but modern membranes are durable and cost-effective. Ultrafiltration, done efficiently, results in little loss of enzyme activity. However, some configurations of apparatus, particularly in which solutions are recycled, can produce sufficient shear to damage some enzymes.
Chapter-10 Concentration and purification of enzyme Concentration by precipitation Precipitation of enzymes is a useful method of concentration and is ideal as an initial step in their purification. It can be used on a large scale and is less affected by the presence of interfering materials than any of the chromatographic methods described later. There are method of salting in and salting out. Salting in is the method in which ammonium sulphate is used for dissolving the protein. Note that protein dissolves least at its isoelectric point. On increasing the ammonium sulphate concentration further proteins starts precipitating this phenomenon is called as salting out. All this process must be done in the ice cold solution so that no protein can denature. Salting out of proteins is done by use of ammonium sulphate, is one of the best known and used methods of purifying and concentrating enzymes, particularly at the laboratory scale. Increases in the ionic strength of the solution cause a
reduction in the repulsive effect of like charges between identical molecules of a protein. It also reduces the forces holding the salvations shell around the protein molecules. When these forces are sufficiently reduced, the protein will precipitate; hydrophobic proteins precipitating at lower salt concentrations than hydrophilic proteins. Ammonium sulphate is convenient and effective because of its high solubility, cheapness, lack of toxicity to most enzymes and its stabilizing effect on some enzymes (see Table 2.4). Its large-scale use, however, is limited as it is corrosive except with stainless steel, it forms dense solutions presenting problems to the collection of the precipitate by centrifugation, and it may release gaseous ammonia, particularly at alkaline pH. The practice of using ammonium sulphate precipitation is more straightforward than the theory. Reproducible results can only be obtained provided the protein concentration, temperature and pH are kept constant. The concentration of the salt needed to precipitate an enzyme will vary with the concentration of the enzyme. However, fractionation of protein mixtures by the stepwise increase in the ionic strength can be a very effective way of partly purifying enzymes. The solubility of an enzyme can be described by the equation (2.11) where S is the enzyme solubility, Kintercept is the intercept constant, Ksalt is the salting out constant and T is the ionic strength which is proportional to the concentration of a precipitating salt. Kintercept is independent of the salt used but depends on the pH, temperature, enzyme and the other components in the solution. Ksalt depends on both the enzyme required and the salt used but is largely independent of other factors. This equation (2.11) may also be used to give the minimum salt concentration necessary before enzyme will start to precipitate; the concentration change necessary to precipitate the enzyme varying according to the magnitude of the salting out constant. Some enzymes do not survive ammonium sulphate precipitation. Other salts may be substituted but the more favored alternative is to use organic solvents such as methanol, ethanol, propan-2-ol and acetone. These act by reducing the dielectric of the medium and consequently reducing the solubility of proteins by favoring protein-protein rather than protein-solvent interactions. Organic solvents are not widely used on a large scale because of their cost, their flammability, and the tendency of proteins to undergo rapid denaturation by these solvents if the temperature is allowed to raise much above 0C. On safety grounds when organic solvents are used, special flameproof laboratory areas are used and temperatures maintained below their flashpoints. Except when enzymes are presented for sale as ammonium sulphate precipitates, the precipitating salt or solvent must be removed. This may be done by dialysis, Ultrafiltration or by using a desalting column of, for instance, Sephadex G-25. A. Nucleic acid removal Intracellular enzyme preparations contain nucleic acids which can give rise to increased viscosity interfering with enzyme purification procedures, in particular ultrafiltration. Some organisms contain sufficient nuclease activity to eliminate this problem but, otherwise, the nucleic acids must be removed by precipitation or degraded by the addition of exogenous nucleases. Ammonium sulphate precipitation can be effective in removing nucleic acids but will remove some protein at the same time. Various more specific precipitants have been used, usually positivelycharged materials which form complexes with the negatively-charged phosphate residues of the nucleic acids. These include, in order of roughly decreasing effectiveness, polyethyleneimine, the cationic detergent cetyltrimethyl ammonium bromide, streptomycin sulphate and protamine sulphate. All of these are expensive and possibly toxic, particularly streptomycin sulphate. Also, they may complex undesirably with certain enzymes. They may be necessary, however, where possible contamination of the enzyme product must be avoided, such as in the preparation of restriction endonucleases. Otherwise, treatment with bovine pancreatic nucleases is probably the most cost-effective method of nucleic acid removal. B. Heat treatment
In many cases, unwanted enzyme activities may be removed by heat treatment. Different enzymes have differing susceptibility to heat denaturation and precipitation. Where the enzyme required is relatively heat-stable this allows its easy and rapid purification in terms of enzymic activity. For such enzymes heattreatment is always considered as an option at an early stage in their purification. This method has been particularly successfully applied to the production of glucose isomerase, where a short incubation at a relatively high temperature is used (e.g. 60 - 25C for 10 min). No interfering activity remains after this treatment and the heat-treated, and hence leaky, cells may be immobilised and used directly. 1. Enzyme purification by Chromatography For identification and separation of biochemical compound chromatography is one of the most effective techniques. The method was developed in 1906 by the Russian botanist Mikhail Tswett. Chromatography involves the principle of partition or distribution coefficient ( Kd ). Kd describes that how a substance distributes itself between different type of immiscible phases. Enzyme preparations that have been clarified and concentrated are now in a suitable state for further purification by chromatography. For enzyme purification there are three principal types of chromatography utilizing the ion-exchange, affinity and gel exclusion properties of the enzyme, usually in that order. Ionexchange and affinity chromatographic methods can both rapidly handle large quantities of crude enzyme but ion-exchange materials are generally cheaper and, therefore, preferred at an earlier stage in the purification where the scale of operation is somewhat greater. Gel exclusion chromatography (also sometimes called 'gel filtration' or just 'gel chromatography' although it does not separate by a filtering mechanism, larger molecules passing more rapidly through the matrix than smaller molecules) is relatively slow and has the least capacity and resolution. It is generally left until last as an important final purification step and also as a method of changing the solution buffer before concentration, finishing and sale. Where sufficient information has been gathered regarding the size and variation of charge with pH of the required enzyme and its major contaminants, a rational purification scheme can be devised. A relatively quick analytical method for obtaining such data utilises a two-dimensional electrophoresis whereby electrophoresis occurs in one direction and a range of pH is produced in the other; movement in the electric field determined by the size and sign of a protein's charge, which both depend on the pH. As the sample is applied across the range of pH, this method produces titration curves (i.e. charge versus pH) for all proteins present. A large effort has been applied to the development of chromatographic matrices suitable for the separation of proteins. The main problem that has had to be overcome is that of ensuring the matrix has sufficiently large surface area available to molecules as large as proteins (i.e. they are macroporous) whilst remaining rigid and incompressible under rapid elution conditions. In addition, matrices must generally be hydrophilic and inert. Although the standard bead diameters of most of these matrices are non-uniform and fairly large (50 150 m), many are now supplied as uniform-sized small beads (e.g. 4 - 6m diameter) which allows their use in very efficient separation processes (high performance liquid chromatography, HPLC), but at exponentially increasing cost with decreasing bead size. Relatively high pressures are needed to operate such columns necessitating specialized equipment and considerable additional expense. They are used only for the small-scale production of expensive enzymes, where a high degree of purity is required (e.g. restriction endonucleases and therapeutic enzymes). Column manufacturers now supply equipment for monitoring and controlling chromatography systems so that it is possible to have automated apparatus which loads the sample, collects fractions and regenerates the column. Such equipment must, of course, have fail-safe devices to protect both column and product.
a. Ion-exchange chromatography Enzymes possess a net charge in solution, dependent upon the pH and their structure and isoelectric point enzyme can be separated. Note that In solutions of pH below their isoelectric point amino acid will be positively charged and bind to cation exchangers whereas in solutions of pH above their isoelectric point amino acid will be negatively charged and can bind to anion exchangers. Therefore pH chosen must be sufficient to maintain a high, but opposite, charge on both protein and ion-exchanger and the ionic strength must be sufficient to maintain the solubility of the protein without the salt being able to successfully compete with the protein for ion-exchange sites. The binding is predominantly reversible and its strength is determined by the pH and ionic strength of the solution and the structures of the enzyme and ion-exchanger. Normally the pH is kept constant and enzymes are eluted by increasing the solution ionic strength. A very wide range of ion-exchange resins, cellulose derivatives and large-pore gels are available for chromatographic use. Ion-exchange materials are generally water insoluble polymers containing cationic or anionic groups. Cation exchange matrices have anionic functional groups such as -SO3-, -OPO3- and -COO- and anion exchange matrices usually contain the cationic tertiary and quaternary ammonium groups, with general formulae -NHR2+ and -NR3+. Proteins become bound by exchange with the associated counter-ions. Ion-exchange polystyrene resins are eminently suitable for large-scale chromatographic use but have low capacities for proteins due to their small pore size. Binding is often strong, due to the resin hydrophobicity, and the conditions needed to elute proteins are generally severe and may be denaturing. Nevertheless such resins are a potential means of concentrating or purifying enzymes. Ion-exchange cellulose and large pore gels are much more generally suitable for enzyme purification and, indeed, many were designed for that task. A variety of charged groups, anionic or cationic, may be introduced. The practical level of substitution of cellulose is limited as derivatisation above one mole per kilogram may lead to dissolution of the cellulose. Consequently, proteins may be eluted from them under mild conditions. Ion-exchange cellulose can be used in both batch and column processes but on a large scale they are used mainly batch wise. This is because the increased speed of large-scale batch wise processing and the avoidance of the deep-bed filtering characteristics of columns outweigh any advantage due to the increase in resolution on columns. Careful preparation before use and regeneration after use is essential for their effective use. Batch wise operations involve stirring the pretreated and equilibrated ionexchanger with the enzyme solution in a suitable cooled vessel. Adsorption to the exchangers is usually rapid (e.g. less than 30 minutes) but some proteins can take far longer to adsorb completely. Stirring is essential but care must be taken not to generate fine particles (fines). Unadsorbed material may be removed in a variety of manners. Basket centrifuges are a particularly convenient means of hastening the removal of the initial supernatant and the elution of the adsorbed material. This is usually done using stepwise increases in ionic strength and/or changes in pH but it is possible to place the exchangers, plus adsorbed material, in a column and elute using a suitable gradient. However, whilst ion-exchange cellulose are widely used for column chromatography on the laboratory scale, their compressibility causes difficulty when attempts are made to use large scale columns. Some of the problems with derivatised cellulose may be overcome using more recently introduced materials. Derivatives of cross-linked agarose (Sepharose CL6B) and of the synthetic polymer Trisacryl have high capacities (up to 150 mg protein ml-1) yet are not significantly compressible. In addition, they do not change volume with pH and ionic strength which allows them to be regenerated without removal from the chromatographic column. More detail description is given in chapter 9. 2 Affinity chromatography This is a term which is based on specific interaction between the enzyme and the
immobilised ligand. In its most specific form, the immobilised ligand is a substrate or competitive inhibitor of the enzyme. Ideally it should be possible to purify an enzyme from a complex mixture in a single step and, indeed, purification factors of up to several thousand-fold has been achieved. An alternative, equally specific approach is to use an antibody (they are specific in binding because they are raised against specific antigen inside an animal) to the enzyme as the ligand. Such specific matrices, though, are very expensive and cannot be generally employed on a large scale. Additionally, they often do not perform as well as might be expected due to non-specific binding effects. In general, affinity chromatography achieves a higher purification factor (with a median value in reported purifications of about ten fold) than ion-exchange chromatography (with a median performance of about three fold), in spite of it generally being used at a later stage in the purification when there is less purification possible. A less specific approach, suitable for many enzymes, is to use analogues of coenzymes, such as NAD+, as the ligand. This method has been used successfully but has now been superceded by the employment of a series of water soluble dye as ligand. These are much cheaper and, usually by trial and error, have been found to have surprising degrees of specificity for a wide range of enzymes. This dyeaffinity chromatography was allegedly discovered by accident, certain enzymes being found to bind to the blue-dyed dextran used, as a molecular weight standard, to calibrate gel exclusion columns. More detail description is given in chapter 9. .3 Hydrophobic interaction chromatography (HIC) or affinity elution HIC is found to be very useful when it was noted that certain proteins were unexpectedly retained on affinity columns containing hydrophobic spacer arms. Hydrophobic adsorbents now available include octyl or phenyl groups. Hydrophobic interactions are strong at high solution ionic strength so samples need not be desalted before application to the adsorbent. Elution is achieved by changing the pH or ionic strength or by modifying the dielectric constant of the eluent using, for instance, ethanediol. A recent introduction is cellulose derivatised to introduce even more hydroxyl groups. This material (Whatman HB1) is designed to interact with proteins by hydrogen bonding. Samples are applied to the matrix in a concentrated (over 50% saturated, > 2M) solution of ammonium sulphate. Proteins are eluted by diluting the ammonium sulphate. This introduces more water which competes with protein for the hydrogen bonding sites. The selectivity of both of these methods is similar to that of fractional precipitation using ammonium sulphate but their resolution may be somewhat improved by their use in chromatographic columns rather than batch wise. CHOICE OF MATRIX Careful choice of matrices for affinity chromatography is necessary. Particles should retain good flow and porosity properties after attachment of the ligand and should not be capable of the non-specific adsorption of proteins. Agarose beads fulfill these criteria and are readily available as ligand supports . Affinity chromatography is not used extensively in the large-scale manufacture of enzymes, primarily because of cost. Doubtless as the relative costs of materials are lowered, and experience in handling these materials is gained, enzyme manufacturers will make increased use of these very powerful techniques. Isoelectric focusing When any enzyme mixture is placed in the gel having solution of different pH then after passing the current a potential difference is established which helps in development of a pH gradient. This pH gradient decides the final movement of the enzyme in the gel, because enzyme will stop its movement only after a situation where pH of enzyme will be equal to the pI of the buffer. (See figure below, enzyme A 1 forming separate bands in compare to the Enzyme A2). Electrophoresis This method was developed by Arne Tiselius in 1937 and is based on principle that any charged molecule will move towards the opposite pole. More detail account is given in chapter 9.
2.11 Maintaining Enzyme Activity The key to maintaining enzyme activity is maintenance of conformation, so preventing unfolding, aggregation and changes in the covalent structure. Three approaches are possible: 1. use of additives, 2. the controlled use of covalent modification, and 3. Enzyme immobilization. In general, proteins are stabilized by increasing their concentration and the ionic strength of their environment. Neutral salts compete with proteins for water and bind to charged groups or dipoles. This may result in the interactions between an enzyme's hydrophobic areas being strengthened causing the enzyme molecules to compress and making them more resistant to thermal unfolding reactions. Not all salts are equally effective in stabilizing hydrophobic interactions, some are much more effective at their destabilization by binding to them and disrupting the localized structure of water (the chaotropic effect, Table 2.4). From this it can be seen why ammonium sulphate and potassium hydrogen phosphate are a powerful enzyme stabilizers whereas sodium thiosulphate and calcium chloride destabilize enzymes. Many enzymes are specifically stabilized by low concentrations of cations which may or may not form part of the active site, for example Ca2+ stabilises amylases and Co2+ stabilises glucose isomerases. At high concentrations (e.g. 20% NaCl), salt discourages microbial growth due to its. osmotic effect. In addition ions can offer some protection against oxidation to groups such as thiols by salting-out the dissolved oxygen from solution. Table 2.4. Effect of ions on enzyme stabilization. increased chaotropic effect Cations Al3+, Ca2+, Mg2+, Li+, Na+, K+, NH4+, (CH3)4N+ Anions SCN-, I-, ClO4-, Br-, Cl-, SO42-, HPO42-, citrate3increased stabilization
________________________________________ Low molecular weight polyols (e.g. glycerol, sorbitol and mannitol) are also useful for stabilizing enzymes, by repressing microbial growth, due to the reduction in the water activity, and by the formation of protective shells which prevent unfolding processes. Glycerol may be used to protect enzymes against denaturation due to ice-crystal formation at sub-zero temperatures. Some hydrophilic polymers (e.g. polyvinyl alcohol, polyvinylpyrrolidone and hydroxypropylcelluloses) stabilise enzymes by a process of compartmentalisation whereby the enzyme-enzyme and enzyme-water interactions are somewhat replaced by less potentially denaturing enzyme-polymer interactions. They may also act by stabilizing the hydrophobic effect within the enzymes. Many specific chemical modifications of amino acid side chains are possible which may (or, more commonly, may not) result in stabilization. A useful example of this is the derivatisation of lysine side chains in proteases with N-carboxyamino acid anhydrides. These form polyaminoacylated enzymes with various degrees of substitution and length of amide-linked side chains. This derivatisation is sufficient to disguise the proteinaceous nature of the protease and prevent autolysis. Important lessons about the molecular basis of thermostability have been learned by comparison of enzymes from mesophilic and thermophilic organisms. A frequently found difference is the increase in the proportion of arginine residues at the expense of lysine and histidine residues. This may be possibly explained by noting that arginine is bidentate and has a higher pKa than lysine or histidine (see Table 2.1). Consequently, it forms stronger salt links with bidentate aspartate and glutamate side chains, resulting in more rigid structures. This observation, among others, has given hope that site-specific mutagenesis may lead to enzymes
with significantly improved stability . In the meantime it remains possible to convert lysine residues to arginine-like groups by reaction with activated urea. It should be noted that enzymes stabilised by making them more rigid usually show lower activity (i.e. Vmax) than the 'natural' enzyme. Enzymes are more stable in the dry state than in solution. Solid enzyme preparations sometimes consist of freeze-dried protein. More usually they are bulked out with inert materials such as starch, lactose, carboxymethylcellulose and other poly-electrolytes which protect the enzyme during a cheaper spray-drying stage. Other materials which are added to enzymes before sale may consist of substrates, thiols to create a reducing environment, antibiotics, benzoic acid esters as preservatives for liquid enzyme preparations, inhibitors of contaminating enzyme activities and chelating agents. Additives of these types must, of course, be compatible with the final use of the enzyme's product.
Chapter-12 Techniques used in Enzyme characterization 2.12 HOW TO KNOW THE PROPERTY OF ENZYME 1. Calculate the molecular weight by following method Ultracentrifugation. Gel filtration. SDS page. Mass spectrometry. 2. Presence of amide or peptide bond can be calculated by UV-IR spectroscopy. Direct structure can be determined by the X-ray crystallography. Amino acid can be determined by Ninhydrin reaction and taking OD at 570 nm. Amino acid sequencing can be done to know the order of amino acid. Introduction After salting in and salting out procedure, enzymes are further purified by two main common techniques 1- Chromatography; 2- Gel electrophoresis. In this chapter we will focus mainly on choice of these techniques and their broad details of the application and principles. A. Chromatography Principles of chromatography For identification and separation of biochemical compound chromatography is one of the most effective techniques. The method was developed in 1906 by the Russian botanist Mikhail Tswett. Chromatography involves the principle of partition or distribution coefficient (Kd). Kd describes that how a substance distributes itself between different type of immiscible phases. Basically, there is several type of chromatographic technique based on following property Adsorption. 2-Ion-exchange; 3-Molecular-sieve Adsorption Chromatography The stationary phase in adsorption chromatography is silica or alumina particles. Analyte are separated due to their varying degree of adsorption onto the solid surfaces. The main advantage of adsorption chromatography is in separating isomers, which can have very different physisorption characteristics due to steric effects in the molecules. Example Thin layer chromatography. Many specialized technique that use them are: --Basically, all type of chromatography consists of two type of phase
Stationary phase: the phase that can not move and is prepared by dissolving some solid , liquid or other material that can be packed inside the column and may provide the support for movement the mobile phase. This may be solid, gel, liquid, or solid liquid mixture. Stationary phase in paper chromatography is the paper, while in ion-exchange chromatography is the ion-exchanger like Dowex -50 , Dowex-1 both are anion exchanger, and the cation exchanger is the CMC carboxymethyl cellulose, DEAE and sephadex. Mobile phase: This is chosen according to the nature of the biomolecules. Mobile phase is used to isolate the biomolecules because most of the biomolecules are present in the solution. Mobile phase is poured in the gel and get separated due to attachment with the stationary phase. The basis of all type of chromatography is the partition or distribution coefficient (Kd). Two immiscible phases are formed after the distribution of compound in the matrix and is denoted by Kd= concentration of compound in phase A / concentration of compound in phase B. 1. Column chromatography is of following type a) GPC or gel permeation chromatography b) Ion Exchange chromatography. c) Affinity chromatography. d) High performance liquid chromatography (HPLC) Based on adsorption of solute in the matrix, adsorption chromatography are of following type a. Thin layer chromatography. b. Paper chromatography. COLUMN CHROMATOGRAPHY In the column chromatography column is filled with the stationary phase attached to suitable matrix and mobile phase passed through the column either with the gravity or by applied pressure. Size exclusion chromatography/ gel filtration (permeation) chromatography History Since the discovery in the 1940s that certain porous material can retain molecules of certain sizes and let others elute, there have been many discoveries that have made size exclusion chromatography practical. Size exclusion chromatography was discovered using starch as a packing material. Although starch was not a very durable material, it led to further interest. Soon, a material called Sephadex, developed for zone electrophoresis, was applied to chromatography; this innovation made size exclusion chromatography realistic. Sephadex is a cross-linked styrene divinylbenzene copolymer, which has led to the development of other divinylbenzene polymers for size exclusion chromatography. The amount of cross-linking (controlled by the availability of divinylbenzene) determines pore size, allowing packing materials to be easily customized for different applications. This initial use of size exclusion was limited to aqueous solvents and called gel filtration chromatography (at Tiseliuss suggestion). The process developed using Sephadex was used extensively in characterizing polymers and polymer molecular weight distributions. When using nonpolar organic solvents; researchers prefer modified divinylbenzene and polyacrylamide as packing materials. Silica has also been used, although the surface is often modified by adding organic compounds to resist adsorption. Today, both gel filtration and gel permeation chromatography are generally referred to as size exclusion chromatography; the processes are still used extensively in polymer chemistry but have gained new uses in biochemistry. Principle Size Exclusion Chromatography differs from conventional chromatography in the behavior of the stationary phase. In most forms of chromatography, the stationary phase chemically interacts through adsorption, charge, or various other actions.
In size exclusion chromatography, the stationary phase simply acts as a sieve to filter molecules based on size. Term gel filtration or exclusion or permeation chromatography is used to describe the separation of molecule of different molecular size utilizing gel material, using molecular sieve. Here separation of molecule is done on the basis of their molecular size and shape with the help of gel that have pores formed during the gel formation. The larger size molecule are separated out or excluded from the pores (see figure), but still they can pass though the spaces around the pore called as void space and appear in the effluent first. Smaller molecules will be distributed between the mobile phases inside and outside the molecular sieve. They will pass through the column at slower rate. Hence they appear last in the effluent. Note that in the column there is always an inert substance. This is the reason why molecules are eluted from the column in order of decreasing size or if the shape is relatively constant. There are various materials that are porous in nature. The most commonly used material is organic gel and they form different type of pores on polymerizations. Therefore, the General principle is simple. The gel is filled in the column that is equilibrium with the mobile phase. Since gel have pores after polymerization and it can be used for the separation of the molecule of fixed size. Kd, denotes the distribution of an analyte (that we have to separate) in a column of a gel is determined solely by the total mobile phase, both inside and outside the gel particle. For given type of gel Kd depends on the molecular size of the analyte. If the analyte is large then kd =0, whereas if the analyte is sufficiently small to gain complete access to the inner mobile phase. So Kd =1. According to availability of the mobile phase kd varies between 0 to 1.
For two substances of different relative molecular mass and kd value for example Kd and kd is the difference in their elution volumes, Vs, can be derived from the equation and shown to be: VS = (Kd- Kd) V1------------------------------------1
The resolution depends on the beads. The coarser beads are unable to hold the fluid, so poorer resolution results. For maximum resolution, superfine beads are used. The gel is a three dimensional network whose structure is usually random. The gels acts as molecular sieves consist of cross linked polymers that are generally inert, do not bind or react with the material that is being analyzed, and are uncharged. The space within the gel is filled with liquid occupies most of the gel volume. The gels currently in use are of three types: ---dextran, agarose, and polyacrylamide. They are used in the aqueous solution.Dextran is a polysaccharide composed of glucose residues. It is commercially available in the trade name Sephadex. They can not be used for molecules that have size more than 600, 000. the alkyl dextran is called as N, N- methylene bis acrylamide. They made strong beads. The product is called as Sephacryl S-300.Agarose is a linear polymer of Dgalactose. It forms a gel held together with crosslink of H-bonds. The pore size is usually much larger than sephadex. This is the reason why they can be used for the separation of the large macromolecular substance like protein, DNA.Polyacrylamide gels are produced by cross-linking acrylamide and N, Nmethylene-bis acrylamide. It is marketed in the name of Bio-Gel P. Mixed gel of
polyacrylamide and agarose is known as Ultra-Gel. Beads can be prepared with defined degrees of porosity, average radii, and mean radii. In general, the porosity of the beads determines the size range of molecules that can be effectively separated- the fractionation range. The radii of the beads are more important for determining the capacity of the column to separate molecules of similar size. Gel permeation chromatography is normally considered to be a medium- to high resolution chromatographic (polishing) technique. Mechanism of separation by size in gel permeation chromatography For our discussion of the theory of gel permeation chromatography, we will make the assumption that many proteins have a roughly spherical shape (often called globular proteins, as opposed to fiber proteins). Access to included volume (volume that hydrates the inside of the bed) depends on the size of the sphere (the hydrodynamic radius) that each protein occupies. The pore size of the bead will determine whether a protein of s Moreover, since proteins have roughly constant density (mass per volume), the size of this sphere is directly related to the mass of the globular protein. Thus, gel permeation chromatography is found to separate globular proteins according to mass. Note, however, that large deviations from spherical shape can cause a protein to migrate anomalously during gel permeation chromatography that is with regard to migration versus mass. Void volume, etc. For chromatography beads of a given porosity, molecules above a certain size will be completely excluded from the pore structure of the beads. These excluded molecules elute in the "void volume" (Vo) of the column and are not fractionated. In contrast, molecules below a certain size for beads of a given porosity will completely penetrate the pore structure and will elute with the "included volume" (Vi). Thus, these molecules are not fractionated either. Only molecules with masses (hydrodynamic radii) that allow them to penetrate the pore structure of the beads only partially fall within the fractionation range and are separated according to their molecular size (approximately, as described above).The use of a gel permeation column as an analytical tool requires the determination of Vo, Vi, and the elution volumes (Ve) of the proteins of interest. By comparing the elution volumes of protein standards of known molecular mass, the molecular mass of an unknown protein can be estimated. In contrast to denaturing gel electrophoresis, which gives subunit molecular masses for an oligomeric protein, gel permeation chromatography provides an estimate of mass for the oligomeric holoprotein. The combination of denaturing gel electrophoresis and gel permeation chromatography is thus exceedingly powerful for elucidating basic features of protein structures. Use of gel permeation chromatography as part of a purification scheme Most protein purification schemes involve several steps. These can be characterized as either bulk or polishing steps. Bulk purification steps are generally useful for samples of low purity and large volume, both of which are typical of the starting material to be used in a protein purification, such as a cellular extract. Common bulk purification methods are centrifugation; precipitation, such as with salt, solvent, acid, or polyethylene glycol; filtration; and ion-exchange chromatography. Polishing purification steps are generally useful for samples of higher quality and smaller volume, typical of partially purified fractions obtained from bulk purification steps. Common polishing purification methods are ion exchange, affinity, and gel permeation chromatography. Gel permeation chromatography is a common and often excellent polishing step during any protein purification; however, it would not be used as a first step in purification except in unusual circumstances (e.g., high abundance of the target protein in the starting material). Table:1 MATERIAL: POLYMER
TRADE NAME
FRACTIONATION
RANGE
1.DEXTRAN < 0.7 1.0-5 1.53.0 4--150 5---600 5---250 10---1500 208000 2. AGAROSE 10-4000 60---20,000 7040,000 A 5m BIO-GEL A 50 SEPHAROSE 4B SEPHYCRYL S G SEPHADEX
G 10 G 25 G 50 G 100 200 200 S 300
S 400 2 B
6B
10---5000 15,000 100---50,000 3. POLYACRYLAMIDE 0.1---1.8 1.06 BIO-GEL
A 15m
40---
P2 P6
P100 5.0--100.0 Overview of a typical gel permeation chromatography experiment In a typical gel permeation experiment, a protein sample (often a partially purified fraction from a previous purification step, see below) is applied onto the top of the column packing (beads) and allowed to flow into them. As additional buffer is applied to the top of the column and allowed to flow through the column, the protein sample migrates through the column. Due to the differential partitioning of molecules into the pore structure of the beads (as described above), the protein molecules are separated. As buffer is eluted from the column, it is collected in fractions until all the components of the original sample have passed through the column.
Note that all the large Ms come out at nearly the same volume, Vo. This is because none of the very large polymers ever enter a pore. So they all elute together at the void volume, Vo. It is customary to plot log(M), not M. Note that the independent variable is plotted on the y axis by convention. With such a curve, you are example, consider point A, you hit the M vs. Ve trend Obtain DRIA similarly from wish! The DRI response is DRI c ( in g/mL) to select a representative sampling of points. For indicated by the cross. Starting at VeA read up until and then read left to get MA from the left y-axis. the right ordinate. Repeat for as many points as you proportional to the concentration of polymer:
The constant of proportionality is dn/dc, the same specific refractive index increment needed in light scattering. You cannot measure concentrations in an isorefractive solvent (i.e., one in which dn/dc = 0). However, it is not necessary to actually know dn/dc in simple GPC. One can obtain average molecular weights without it. For example: Mw = =
Since, the constant of proportionality factors out of the numerator and denominator identically. One can also obtain the number average molecular weight: Mn = = = =
ADVANTAGE OF GEL CHROMATOGRAPHY In this chromatography the material used is inert one so there is no effect of temp. pH, ionic strength. They can be used for very labile material like enzyme. Since there is no adsorption by the inert material so labile material are not affected. The elution volume is related in a simple manner to molecular weight. Application of GPC or gel permeation chromatography Size exclusion chromatography is used primarily in two areas of chemistry: polymer chemistry and biochemistry. In polymer chemistry, size exclusion chromatography
can separate polymers with different numbers of monomer units, giving the polymers producer information about the length of the chains produced. In addition, because size exclusion chromatography does not destroy or alter the sample, the fractions of polymer can be further tested. For example, S. J. ODonohue and E. Meehan discuss the use of different solvents in size exclusion chromatography and molecular weight detectors (light scattering and viscometry) at high temperatures to characterize polymers, such as o-chloronaphthalene, that can be processed only at temperatures above 135 C (1999). Antonio Moroni and Trevor Havard discuss the use of solvents in the analysis of engineering thermoplastics, particularly polyesters and polyamides (1999). Their experimentation leads to the suggestion that 1,1,1,3,3,3-hexafluoro-2-propanol may not be a good solvent for size exclusion chromatography as polar portions of macromolecules (such as polyesters and polyamides) may be solvated and extended, making these molecules seem larger than they actually are. This extension or enlargement causes them to be eluded at later times than expected. In biochemistry, size exclusion chromatography is useful in separating highmolecular-weight molecules from other molecules. For example, in a genetics study, Anke van Rijk and colleagues used size exclusion chromatography to isolate a small protein called alpha-A-crystalline to study the insertion of hamster alpha-Acrystalline into mice (1999). size exclusion chromatography allows the researchers to isolate the biomacromolecule based on its relative size without destroying the sample. Biochemical separations may seem to make this more of a separation technique, but in both polymer chemistry and biochemistry, size and molecular weight estimations can be made. If a polymer chain of unknown size, or a protein of unknown size, is analyzed along with molecules of known size or molecular weight, then when the compound in question eludes, it can be compared with the molecules that it eluded closest to determine an approximate molecular weight. In purification of Macromolecule: GPC can be used to separate the viruses, proteins, hormones, enzyme, antibodies, nucleic acid and polysaccharide. In study of protein binding study Estimation of the molecular weight: Gel 1 gel 2 Log M THE plot is between K verses log M (molecular weight). It yields a straight line except for very small and very large molecule. The parameter K = Ve---Vo / Vs, where Ve= elution volume, Vo= void volume, Vs= volume of stationary phase or = Vt---Vo, Vt is the total volume of the column PAPER CHROMATOGRAPHY Cellulose has many hydroxyl groups which are polar and bind H2O. The bound H2O is the Stationary Phase. H2O run across the cellulose paper due to capillary action. Mobile Phase is mixture of organic solvents (alcohols, ketone, aldehyde etc.) and possibly water. The mobile phase solvent will be less polar than H2O; it is usually a mixture of Solutes partition between H2O in the stationary phase and the solvent of the mobile phase. One can change the characteristics of separation by changing the polarity of the mobile phase (i.e. adjust the composition). Paper Chromatography is the most common form of cellulose chromatography in which solute is "spotted" on "dry" paper (still contains H2O) encircle by the pencil at a line mark and chromatograph is "developed by dipping one end in the mobile phase. There are two modes of chromatography1: Ascending and 2-Descending.The solvent moves through the paper, drawn by capillary action Solutes move as spots with a rate depending upon how much time they spend in the stationary phase vs. the mobile phase--determined by their partition coefficient--measured as an Rf
value. Maximum distance covered by the solvent is noted and the distance covered by the solute is noted. The ratio of two gives Rf value. This Rf value is fixed and is generally less than one. Ion exchange chromatography This type of chromatography is used for many biological materials like amino cid and proteins, which have ionisable groups, i.e. they have either negative or positive charge. The name ion exchange is given because of exchanging ions for ion in aqueous solution. The principle of ion exchange chromatography is that charged molecule adsorb to ion exchangers reversibly so that molecule can be bound or eluted by changing the ionic environment. Separation by ion exchange is usually involving two step. First the substances to be separated are bound to the exchanger, using the conditions that make them more stable and tight binding. Ion exchange separation is carried out mainly in column packed with an ion exchangers. The column packing for ion chromatography consist of ion-exchange resins bonded to inert polymeric particles (typically 10 m diameter). For cation separation the cation-exchange resin is usually a sulfonic or carboxylic acid, and for anion separation the anion-exchange resin is usually a quaternary ammonium group. For cation-exchange with a sulfonic acid group the reaction is: -SO3- H+(s) + Mx+(aq) -SO3- Mx+(s) + H+(aq) where Mx+ is a cation of charge x, (s) indicates the solid or stationary phase, and (aq) indicates the aqueous or mobile phase. The equilibrium constant for this reaction is: [-SO3- Mx+]s [H+]aq Keq = -----------------------------------------[-SO3- H+]s [Mx+]aq Different cations have different values of Keq and are therefore retained on the column for different lengths of time. The time at which a given cation elutes from the column can be controlled by adjusting the pH ([H+]aq). Most ion-chromatography instruments use two mobile phase reservoirs containing buffers of different pH, and a programmable pump that can change the pH of the mobile phase during the separation.
There are two type of ion exchanger 1) cationic exchanger 2) Anion exchanger. Cationic exchanger posses negative charge, while anionic exchanger posses positive charge. The negative charge ion exchangers bind to positive charge molecule. The more highly charged molecule to be exchanged, the tighter it binds to the exchanger and less readily it is displaced by other ion. A typical group used in ion exchange is the sulphonic group, SO3 -. If an H+ is bound to group, the exchanger is said to be in the acid form. It can exchange one H+ for one Na+ or two H+ for one Ca++. The sulphonic acid groups are called as a strongly acidic cation exchanger. Other weakly acidic cation exchangers are phenolic hydroxyl group and carboxylic group. Strong Ion Exchangers: based upon strong acids or bases and are charged over a wide range of pH a. strong cation exchangers: Sulfonic Acid (or derivatives) RSO3- b.strong anion exchangers: quaternary ammonium salts R-N (CH3)+ Weak Ion Exchangers: based upon weak acids or bases which are charged only over a limited pH range a. weak anion exchangers: Diethyl-amino-ethyl (DEAE); tertiary amines b.weak cation exchangers: carboxy methyl (CM); phosphoryl. These may be bonded to a variety of supports: e.g. DEAE-cellulose; DEAE-Sephadex; DEAE-Sepharose; CMCellulose; CM-Sephadex; CM-Sepharose Choosing an Ion Exchanger Charge -- cation or anion exchanger -- depends upon the charge of the molecules to be separated. This will also depend upon pH. Weak Exchanger is useful for labile
molecules such as proteins. Strong Exchanger is used for more stable molecules such as nucleotides, amino acids, peptides etc.The matrix is used of various materials like dextran, agarose, cellulose and copolymer of styrene and vinyl benzene. The total capacity of ion exchanger is measure of its ability to take up exchangeable ion. It is measured in term of milliequivalent of exchangeable group permiligram of dry weight. The available capacity is the capacity under particular conditions like pH, ionic strength. The extent to which an ion exchanger is charged depends upon the pH. The porosity of the matrix is very important factor because of charged group are present both outside and inside of the matrix. Since pore also act like molecular sieve. Large molecules may be unable to penetrate the pore so the capacity will decrease with increase in molecular weight. Ion exchanger come in variety of particle size called as mesh size. Finer mesh means an increased surface to volume ratio and therefore increased capacity and decrease time for exchange to occur for given volume of exchanger. Finer the mesh the slower will be the flow rate. For materials that have either single charge the choice of exchanger is not difficult. But in case of material that have both negative and positive charge the choice depends upon the charge that is stable. As we know above the isoelectric point if the pH is stable one then an ion exchanger can be used. If stable below isoelectric point then a cation exchanger can be used. If the substance is labile, a weak exchanger can be used. The sephadex and bio-gel exchanger offers a particular advantage for macromolecules that are unstable in low ionic strength. Because the cross links in these materials maintain the insolubility of the matrix even cross link in these materials maintain the insolubility of the matrix even if the matrix is highly polar. Macromolecular separation always needs larger pore size while micro molecule can be separated by the small pore. It is because of larger available capacity. The cellulose ion exchanger proved to be best for the macromolecule like protein and the nucleic acid. Small mesh sizes improve the resolution but decrease flow rate, which increases the zone spreading and decrease resolution. For selection of buffer following rule is always adopted. The cationic buffer is used with the anionic exchanger, and anionic buffer is used with the cationic exchanger. Simply because, the binding strength, depends on the ionic factor. For best ionic resolution ionic condition should be same as the eluting condition. Eluting Ion Exchange Columns -- molecules usually adsorb tightly in the column so, this interaction, must be weakened. (Most common method): F = q1 q2 / D r2 q1and q2 are the charges on two groups, r is the distance between the groups, and D is the dielectric constant of the solvent which is increased with higher ionic strength thus weakening the force between the solute and the ion exchanger. Another way to look at this is that other ions in the buffer compete for the ion exchanger binding site.change pH -- changes the charges on the molecules being separated; also can change the charge of a weak ion exchanger .These changes can be made stepwise by changing the buffer reservoir (step gradient) or as gradient -- by mixing two buffers The basic principle of ion exchange chromatography is that the affinity of substance for the exchanger depends on both the electrical properties of the material and the relative affinity of other charged substances in the solvent. Hence, bound material can be eluted by changing pH, thus altering the charge of the material. In deciding the eluting condition, it is important to consider how eluting fluid will affect the assay for the material. For example if spectral analysis is to be done then eluting fluid should not absorb in the required wavelength. One important use of on exchange is in desalting. Gel chromatography is an effective means of removing ion from solutions of macromolecules. A related resin called a mixed bed resin is used to prepare deionized water. Ion exchanger has been found to be an effective way to separate weakly polar substances by using the exchanger as the matrix for partition chromatography. For example to separate the sugar mixed with the polar and non-
polar solvent, the solution is applied to the column. The polar solvent binds to the matrix forming the polar stationary phase and sugar partition between this phase and the mobile weakly phase. This is called as reverse phase chromatography. Gas / Liquid Chromatography In gas chromatography mobile Phase is usually a gas -- usually inert (He, Ar, N2) and Stationary Phase is a liquid coating on an inert solid support. There is open tube or capillary operation and other a coat the inside surface of a long thin tube (30 - 100 m long) Packed column -- larger diameter column is packed with an inert support -commonly diatomaceous earth, teflon powder, or glass beads . Coating may be solid at room temp (e.g. polyethylene glycol) but the column is run at higher temperature where the coating melts. Sample usually injected as a liquid which is then heated to vaporize it. Note that sample must be somewhat volatile and stable at higher temperature. GC Can provide very high resolution by making very long columns. Detectors present are of various types. Most powerful detector is a mass spectrometer (GC/Mass Spec.) which give mass spectrum that can be used to identify and quantitate samples as them come off the column. Reversed Phase Chromatography In reverse phase chromatography stationary Phase is apolar (hydrophobic) and is reversed with respect to cellulose chromatography. This hydrocarbon chains is bound to an inert matrix to provide hydrophobicity. Hydrophobicity can be varied by changing the hydrocarbon chain length or by aromatic groups. Other phase is mobile Phase that depends upon hydrophobicity of stationary phase. Commonly use a more polar organic solvent like acetonitrile, DMSO, EtOH, ethylene glycol, propanol, or mixtures of these with H2O. Use of shorter hydrocarbon chains less densely packed is called Hydrophobic Interaction Chromatography HIGH PRESSURE LIQUID CHROMATOGRAPHY [HPLC] Conventional liquid chromatography uses plastic or glass columns that can range from a few centimeters to several meters. The most common lengths are 10-100 cm, with the longer columns finding use for preparative-scale separations. Highperformance liquid chromatography (HPLC) columns are stainless steel tubes, typically of 10-30 cm in length and 3-5 mm inner diameter. Short, fast analytical columns, and guard columns, which are placed before an analytical column to trap junk and extend the lifetime of the Stationary Phases. Usually the resolving power of a column increases with the length of the column and the number of theoretical plates per unit. The number of plates increases as the available surface area per unit length of the column becomes greater in other words as the matrix particle become smaller. However, the flow rate of a column drops as the particle size decreases and this allow sufficient time for significant diffusional spreading to occur. This decreases the resolution. Diffusional spreading can be reduced if the transit time of the mobile phase in the column made small. This can be done by establishing a pressure difference across the top and bottom of the column to force the liquid through the bed. The use of very fine particle and very high pressure to maintain adequate flow rate is called as HPLC or high performance or high pressure liquid chromatography. HPLC is known for its rapid separation with extraordinary resolution of peaks. Also it requires the less amount of test material. It is because of its small fraction of the void volume. For e.g. with a typical column having a diameter of 36mm and a length of 10-20 cm operating at 50-200 pounds per square inches [ 520 kg/ cm2 ] , a sample of 0.01 to 0.1 ml. can be used. The pressure used affects the resolution of the bands. At atmospheric pressure diffusion spreading causes the bands to overlap. However, if pressure (and hence flow rate is very high, there may be insufficient time for molecules in the mobile phase equilibrate and the bands are also broadened. At present time this procedure is applied principally with the ion-exchange and adsorption chromatography of small molecule, peptide, small carbohydrate and t-RNA.
Partition Chromatography In partition chromatography the stationary phase is bonded to inert particles of 3-10 m of diameter, with the smaller sizes, 3-5 m, being used in analytical columns, and the larger particles being used in preparative-scale HPLC. Analyte separate as they travel through the column due to the differences in their partitioning between the mobile phase and the stationary phase. Reverse-phase partition chromatography uses a relatively nonpolar stationary phase and a polar mobile phase, such as methanol, acetonitrile, water, or mixtures of these solvents. The most common bonded phases are n-octyldecyl (C18) and n-decyl (C8) chains, and phenyl groups. Reverse-phase chromatography is the most common form of liquid chromatography, primarily due to the wide range on analyte that can dissolve in the mobile phase. Normal-phase partition chromatography uses a polar stationary phase and a nonpolar organic solvent, such as n-hexane, methylene chloride, or chloroform, as the mobile phase. The stationary phase is a bonded siloxane with a polar functional group. The most common functional groups in order of increasing polarity are: cyano: -C2H4CN diol: -C3H6OCH2CHOHCH2OH amino: -C3H6NH2 dimethylamino: -C3H6N(CH3)2 Chromatographic Separation principle Commercial name Phase Type of support Adsorption Partisil C8 Corasil Pellumina Partisil Micro Pak Al Bondapak C18 ULTRA pak TSK ODS Octylsilane Silica Alumina Silica Alumina Porous Pellicular Pellicular Microporous Microporous Pellicular Porous Ion exchange Partisil-SAX MicroPak-NH2 Strong base Weak base Porous Porous Exclusion Styragel Superpose Bio-glass Nature of stationary
Fractogel TSK Glass Polystyrene-divenyl benzene Agarose Polyvinylchloride Rigid solid Semi-rigid gel Soft-gel Semi-rigid gel Affinity chromatography Principle Affinity chromatography is almost exclusively used for the purification of biological molecules such as proteins and other macromolecules. The technique has been known for almost a century, but suitable support materials were not available until much more recently. With new materials and new demands, the technique became extremely useful in the 1960s, and it has become essential with the growing demand of the biotech in the 1980s and 1990s.Biotech research has used affinity chromatography because of its ability to separate one desired species from a host of other biological molecules. Specificity based on three aspects of affinitythe matrix, the ligand, and the attachment of the ligand to the matrixis the hallmark of this process and the reason for its success. As the name suggests, it utilizes the property of biological affinity of the substances to be separated. As a consequence, it is capable of giving absolute purification, even from complex mixtures, in a single process. Affinity chromatography operates on the principle that ligand (as stated in the Table), attached to a matrix made up of an inert substance, bind to the desired molecule within a solution to be analyzed . Ideally, the ligand will interact only with the desired molecule and form a permanent bond. All other compounds in the solution will elude, leaving the desired product in the column. The desired molecule is then removed from the column by using a wash (typically changing the pH) that lowers the dissociation constant and allows recovery of a nearly pure sample. Choosing the correct ligand is the first hurdle. The ligand must bind strongly with the molecule that is to be recovered. Biological systems have millions of ligand (also known as receptors), and most can be affixed to the matrix and used to isolate the desired molecule. If the ligand chosen can bind to more than one molecule in the sample in question, then a technique called negative affinity, which uses ligand to remove everything but the target molecule from the solution, may be used. For example, if you were looking for a molecule from a cell, but the only ligand that binds the molecule also trapped two additional, different molecules, you could run a normal affinity column and collect these three molecules. Then, if a ligand were found that attached to the two unwanted molecules, that ligand could be used in the size exclusion chromatography and affinity column, allowing the desired molecule to elute while the other two were retained in the column. Matrix materials simply hold the active ligand and provide a pore structure to increase the surface area to which the molecules can bind. Ligand attachment requires that the matrix be activated and then react with the ligand to fix them onto the matrix. During this process, the ligand must also remain active toward the target molecule, or all will be for naught. Substituent groups within the matrix, such as amino, hydroxyl, carbonyl, and thio groups, are easily activated and can serve as the sites to which the ligand attach. Matrix materials are often polysaccharides, such as agarose, that have many hydroxyl groups that can be activated. The matrix, in addition to requiring activation, must also often stand up to decontamination when purifying pharmaceutical compounds. Decontamination is typically performed by rinsing the column with sodium hydroxide or urea. Different matrix materials are stable in different pH ranges, adding the third aspect of selection for affinity
chromatography. The technique was originally developed for the purification of enzymes, but it has been extended to nucleotides, nucleic acids, immunoglobulin, and membrane receptors and even to whole cells and cell fragments. The technique can be shown by the following equation: if we assume M is the macromolecule and L ligand is attached to the matrix, then an ML complex will be formed. M + L ML
For the success of the experiment following steps must be kept in mind. 1. Matrix should have broad range of thermal and chemical stability. It should not absorb any chemical or substance to be purified itself. 2. The ligand should be coupled without altering its binding properties. 3. A ligand must bind tightly. 4. During the elution the matrix should not be destroyed. The most useful material is the Agarose, and polyacrylamide, since they have broad range of thermal tolerance, exhibit minimum absorption, maintain good flow property after coupling, and does not denature after application of extreme pH and ionic strength. The choice of ligand depends on the specificity of the substance. For example for an enzyme ligand must be the substrate, a reversible inhibitor, or an allosteric activator. METHOD OF LIGAND IMMOBILIZATION Linking or coupling of the ligand to matrix material is called as immobilization. 1. CNBR activated agarose CNBr is negatively charged so they can react strongly with the amino group. It is extremely useful for coupling enzyme, Co-enzyme, inhibitors, antigen, antibody, nucleic acid and most protein to agarose. 2. 6-AMINO-HEXENOIC ACID & 1,6 DIAMINO HEXANE. a) Used mainly for small ligand. b) They can solve the steric problem c) Only a Spacer can be attached between matrix and ligand. d) Spacer can be attached by reacting functional group C00H (by using Agarose) and NH2 (group of 6AHA). e) Epoxy activated Agarose.This is useful for linkage of sugars and carbohydrate or any material containing OH gp, amino gp, thiol gp. f) Thiopropyl-Agarose.Can be Used or sulpher containing protein. Before using, it should be treated with the cysteine. 3. Carbomyl diimidazole activated agarose. Coupling of N-nucleophile to CNBr. This results in isourea linkage that carries potential charge and thus can act as an ion-exchanger. Table9. 2 Affinity chromatography Substance Use Lectin Polysaccharides and glycoprotein present on the RBC membrane. Con A Sepharose They selectively can binds to the N-acetyl glucosamine residue. Therefore they can be used for the lymphocyte separation. Helix pomatia lectin Used for separation of the pure T-cell Lactose Caster bean, pea nut can be used for separation of lactose. Maltose Can be separated by the jack bean. D-glucosamine Can be separated by clam
NADP+ Can be separated by using 2, 5 ADP. Protein A Can be used for separation of IgG (using Fc region) Poly A Can be separated of m-RNA Boronate polyacrylamide RNA, sugar, catecholamine Heparin-Agarose Blood protein, DNA polymerase, Ribosome, androgen receptor Imino-diacetic acid Zn+2, Cu +2 Can be separated Octyl agarose Protein Can be separated Thio-propul Sepharose Can be separated by clam Protein, Urease, and Papain. Applications The applications of affinity chromatography have been numerous, but they are predominantly in the field of biochemistry. Early applications were in the separation of biomacromolecules from other biological compounds, such as molecules from an entire cell. This continues to be the primary and extremely important field for affinity chromatography. For example, S. Loukas and colleagues studied the existence of one type of suspected opiate receptor in the brain (1994). They separated the ( -opioid binding protein by using an opioid receptor antagonist that was specific to the ( -opioid binding protein. These studies may one day lead to a better understanding of how drugs such as opium affect our brains. In addition to using small ligand to separate large molecules, researchers have immobilized large molecules on the matrix and used them to separate the small molecules that bind to them. In addition to biological separation, affinity chromatography can be used to determine dissociation constants of ligand and molecules. The longer a molecule stays on the column, the broader the chromatographic peak, indicating that the molecule is more tightly bound to the ligand. This quantitative information can be used in further studies of the ligand or molecule. Thin layer chromatography Principle in this type of chromatography the stationary phase is generally the glass plastic or metal foil plate. The mobile liquid phase passed through the thin layer plate. The layer is formed must be thin Mobile phase is passed through the stationary phase under capillary force, and the analyte is distributed well in the layer. The distribution coefficient is represented by the Kd. Distribution process is based on the fact of adsorption, partition, ion-exchange, exclusion chromatography. The movement of analyte is expressed by the retardation factor. Rf= origin. K can be calculated by = 1Rf/ Rf Process of preparation of the thin layer plate A thin layer of about .25 mm thick slurry is prepared in water is applied to glass with the help of plate spreader. CaSO4 is mixed to facilitate adhesion of the adsorbent to the plate. The plate is dried at 1001200C. This is also helpful in the activation of the plate. Sample is applied to the plate 22.5 cm from edge by means of micropipette or microsyringe. The TLC plate is dipped in the developing phase to depth of about 1.5 cm it is left for one hour. Detection is done by the several methods. For e.g. fluorescent dye (this can be Distance moved by the solute from origin / Distance moved by the solvent from
absorbed by the UV light. For investigating the unsaturated compound I2 vapour can be used. For radiolabel led compound autoradiography can be used. Movement of compound can be observed by the specific Rf values. ADVANTAGE OF TLC OVER OTHER CHROMATOGRAPHY TECHNIQUE IT has great resolving power and greater speed of separation. A wider range of sorbant can be used and also due to easy detection of spot and separation of chromatogram. Two factor that affect the TLC has, High surface to volume ratio and the weight ratio, If necessary 2D gels electrophoresis can be used in the TLC plate this can be used by placing the plate in the buffer at right angle to first separation. Commonly used separating agent is the Ninhydrin for detection of the amino acid. Rhodamine B is used for the detection of the lipid; SbCl2 for steroid and tarpenoid; H2SO4 for organic acid, H2SO4 and KMN03 for hydrocarbon. Br2 vapour is used for olefins detection. HPTLC and HPPLC Thin-layer chromatography (TLC) gets the high-performance (HP) treatment through several optimization steps leading to improved efficiency and automation. The layers are made smaller (0.02 mm instead of 0.25 mm), they have smaller mean grain sizes of the coating particles (down to 7 m instead of 1230 m), and the grain size is more uniform. Equally if not more important, HPTLC shows better optical properties than standard TLC, allowing for easy densitometry studies. Better resolution as well as a 10-fold improvement in detection limits are key to HPTLCs increasing popularity. By using a forced-flow mobile phase and a sealing chamber, researchers can transform HPTLC into high-pressure planar liquid chromatography (HPPLC), gaining the added benefits that pressure provides, especially to the speed of the run. HPTLC can also be used with centrifugation to force the sample outward in what is known as rotational planar chromatography (RPC). These adaptations are collectively responsible for what is known as modern TLC, which relies heavily on instrumental analysis. Applications of these modern forms are extensive, including routine use in the pharmaceutical industry and clinical analysis, in food analysis, natural products analysis (including a variety of botanicals and herbals), and in environmental analysis for determining such things as pesticides in drinking water. Selection of chromatographic system i. Ligand having specific binding propertyAffinity chromatography. ii. Volatile compound ----gas liquid chromatography. iii. For compound having different functional groupadsorption chromatography. iv. Low polar compound---liquid chromatography. v. Water soluble but weakly ionic---reverse phase liquid chromatography. vi. Water soluble but strong ionic compound---ion-exchange chromatography. vii. Compound differ in molecular sizeexclusion chromatography. Electrophoretic technique Electrophoresis, migration due to an electric field, was first discussed by L. Michaelis (1909). Arne Tiselius rediscovered this technique in 1937 and used it to separate human serum proteins. In subsequent years, development focused on new gel materials that minimized convection, which interferes with charged species migration. In the 1960s, polyacrylamide gels were developed, which ushered in the widespread use of gels in biological studies. Capillary electrophoresis was pioneered by S. Hjerten, who built tube electrophoresis units as early as 1959.However, the technology on which this technique depends, such as high-quality capillary silica tubing and extremely sensitive detectors, had yet to mature. It took two more decades before Jorgen son and Lukacs developed the first truly useful CE instrument and showed that it had exceptional resolution (1981). Theory. CE uses the phenomenon of electroosmotic flow to separate analytes. CE
instruments consist of a capillary column between two reservoirs of buffer solution (the solution used as the solvent). Electrodes are placed in both reservoirs, which places a potential across the column. The column itself becomes negative if the cathode is on the product side and the anode is on the sample side. When the column is negative, cations migrate toward the column wall, creating a dense +ve area. This area is then attracted by the cathode and starts to flow toward the product end of the column. These cations pull the rest of the fluid with them. A reversed-phase HPLC packing material is used to retain the analytes at different rates on the liquid stationary phase. Traditional CE uses polar solvents, precluding the identification and analysis of polar compounds. But in the early 1980s, Terabe and colleagues developed a variant of CE that allows determination of nonpolar molecules (1984, 1985). Micellar electrokinetic capillary chromatography (MEKC) uses surfactants or polymers to form micellessmall spheres with hydrophobic centers and ionic shields around the outsidethat travel against the liquid flow toward the anode (the charges could be switched with different surfactants) but are swept with the electroosmotic flow toward the cathode. However, they will elute last because of their struggle to reach the anode. Hydrophobic molecules will enter the micelle, but they will also exit in a fashion similar to liquid partition chromatography. Therefore, the separation of molecules is based on their polarity as polar molecules are slowed by entering the micelle. It describes the migration of a charged particle under the influence of electric field. Many important biomolecules can be separated by this technique like: amino acid, peptide, protein, nucleotide, ionisable group. The gel is prepared from the material like agarose or polyacrylamide and the separation of compound is done in the buffer. The substances separated at the low temperature to save it from heating effect. The function of buffer is to maintain the constant ionization pattern of the molecule. Force can be represented by the following equation: F= qv/ velocity q-= charge and V is the potential difference. Electrophoretic mobility () is defined as ratio of velocity to field strength.
a. Agarose Gel Electrophoresis Agarose is prepared from natural compound isolated from Gracilaria and Geladium. The basic repeat unit is the galactose and 3,6- anhydro galactose. Generally the agarose concentration is used in the 1% to 3% . Agarose has property to dissolve in the hot water making liquid solution while it solidifies in the cooled medium and forms a rigid gel. The gel is formed due to inter and intra-H bonding. This cross linking attaches new anti conventional properties. The pore size is decided by the initial concentration of the agarose. High concentration of agarose gives usually smaller pore size. While, small concentration of agarose gives larger pore size. The main problems occurs during electrophoresis is the phenomenon of electro endosmosis. This can be avoided by the substituting some other functional group like sulphate. 1% Agar is used for the immuno-elctrophoresis or flat bed electrophoresis. Generally pore size of the 1% Agar has pore size larger than proteins and nucleic acid. Agar can be used more for analysis of DNA. For this purpose vertical slab gel can be used. b. Polyacrylamide Gel Electrophoresis After the polymerization of acrylamide it forms a polymer compound called as polyacrylamide. And so electrophoresis is called as polyacrylamide gel electrophoresis or PAGE. THERE is some necessary component for the polymerizations of the acrylamide like the N, N bis acrylamide. Note that bis acrylamide is
essentially two acrylamide linked by the methylene group. Initiation of the polyacrylamide is difficult so some more compound is added like ammonium persulphate, and the TEMED or N,N,NN tetramethylenediamine. Actually addition of TEMED IS necessary as it acts like the catalyst. S2O8 + eSO4 2+ SO4
IF this free radical is represented as R. Oxygen can be used for the removal of the free radicals. Free radical generation can be done by the photo polymerization. Low percentage gel can be used for the separation of the DNA. For protein separation SDS PAGE can be used the usual percentage lies between the 3 to 30 % of acrylamide. Since protein separation done in the two steps: first it is separated in the stalking gel, and size exclusion chromatography on it is resolved by the resolving gel. Stalking gel generally uses lower percentage of the gel that provides the larger pore size. It allows the free movement of the protein so that one type of protein either same in the molecular weight or charge can stalk at one position. After this process the protein is separated in the gel having smaller pore size. All these process can be done by the vertical slabs. c. SDS-PAGE SDS is an anionic detergent (CH3(CH2)10 CH2OSO3-Na+). With mercaptoethanol and SDS, protein can be denatured into the simple protein. It helps in the better separation of the protein. And thus protein is analyzed in qualitative way. On average it has seen that one SDS can binds to the two amino acid. Problem arises during the electrophoresis is the settling of sample and exact tracking of the protein position in the gel. For settling problem sucrose solution is used, and for tracking the protein bromo-phenol blue is used. The protein first of all loaded in the stalking gel. In this master mix, glycine is added. This is done to sharpen the protein band. But note that glycinate ion have a lower electrophoretic mobility than protein SDS complex, which have lower mobility than chloride ion of the loading buffer and the stacking gel Cl- > PROTEIN-SDS > GLYCINATE So protein SDS band lies in between the Cl- and glycinate ion. PH of the stacking gel is generally 6.8 and resolving gel has pH about 8.8. The negatively charged protein is attracted toward the anode. After the protein is reaches the bottom, the gel is removed and placed in stain more generally the Coomassie blue. The gel is then placed in the destain solution. Staining of gel is done for 2-3 hrs. And destaining requires overnight. Generally 15 % polyacrylamide gel is used in the separating gel. This can allow separation of the protein in the range of 100,000 to 10, 000. For protein of molecular weight more than 150, 000 7.5% gel is used. Molecular weight of protein Mr can be determined by the comparing its mobility with those of a number of protein used as standard. By plotting a graph of distance moved against log Mr for each of standard proteins, a calibration curve can be constructed. The distance moved by the protein of unknown Mr is then measured and its log Mr and hence Mr can be determined from calibration curve. A protein generally have single band in the protein unless it has two same subunit. d. Native (buffer) gel In this method SDS is not used for the separation of the protein subunit prior to loading. Here protein is separated according to the native charge of protein at pH of the gel (normally pH 8.7) and according to the different electrophoretic nobilities and the sieving effect of the gel. The method is generally used for the enzyme substrate reaction and total protein content. e. Gradient gel The protein is separated according to the gradient formed by the different concentration of the gel. Generally 5% at the top, while 15% gel is used at the
bottom. The main advantage is that very similar molecular weight can be resolved. f. Iso-electric focusing: IEF gel: It utilizes the horizontal gels and separates the protein according to the isoelectric point of the protein. As we know that protein is the ampholytes, it can be separated easily. Here riboflavin is used for initiation of the polymerization of the gel. Protein having pH lower than iso-electric point will be positively charged, and will initially migrate towards the cathode. As they proceed further, the charge on the protein decreases slowly and at one point protein stops where IP is equal to the charge. For determining the IP of the protein, a standard IP of known protein can be determined and with the help of this calibration curve, IP of unknown protein can be determined. IEF is highly sensitive technique and used for studying heterogeneity of the protein. g. Chromatofocussing Is suitable for the protein separation. This is based on forming a pH gradient. The ion-exchanger is fixed in the column and set to a particular temperature and pH. The difference of pH lies between 3-4 unit from upper and bottom. On adding protein at top of column the protein moves at its isoelectric point. Just beyond the isoelectric point the protein is bind to the positive charge of the ionexchange column. Chromatofocussing gives a good resolution of quit complex mixture of protein provided that there is close difference in their isoelectric point, it gives poor resolution of compound having same isoelectric point.
2D-PAGE This technique utilizes the technique of both IEF and the SDS-PAGE. In the first dimension isoelectric focusing is done in polyacrylamide gel in narrow tubes in the presence of ampholytes, 8M urea and non-ionic detergent. Denatured protein is separated in the gel according to the isoelectric point. In another step the protein is run in presence of SDS. Generally the technique is used in the translational product of the gene or mRNA. It also helps in isolating the extra protein in the expression. Now a days to reduce so much of the labor, computerized 2D is done. DETECTION, ESTIMATION AND RECOVERY OF PROTEIN GELS. CBB or Coomassie blue R -250 is most commonly used for the detection of the protein in the gel. Staining is done in the 0.1% (w/v) CBB in methanol: water: glacial acetic acid. This acid methanol mixture acts as a denaturant to precipitate or fix the protein in the gel. This prevents the protein from being washed out while it is stained. Coomassie blue is highly sensitive, means it can detects the 0.1 g of protein. Silver stain is more sensitive. Ag+ ion is reduced to metallic silver on the protein, where the silver is deposited to give black band. It can be used immediately after the electrophoresis. It is 100 time more sensitive than CBB. Glycoprotein can be detected by the stain called as PAS (periodic acid stiff stain). It gives pink red bands. It is difficult to observe in the gel. Quantitative analysis of the protein can be done by the method called as scanning densitometry. Protein can be further analyzed and protein can be purified. Protein band can be cut out of protein and sequence by the gas phase sequencer. Note: separation of protein is called as western blotting, while separation of protein is called as southern blotting. i. AGAROSE GEL ELECTROPHORESIS OF DNA As we know that DNA is larger than proteins and therefore they are unable to enter the polyacrylamide gel. So the convenient way is to use agarose to analyze the DNA. Note that single stranded DNA is always expressed in nt (nucleotide) while double stranded DNA is expressed always in the base pair or kilo base pair. Agarose gel of 0.3% will separated double stranded DNA molecules of about 5 and 60
kb size, whereas 2% gels can be used for the separation of the sample between 0.1 and 3 kb. Most of the lab uses 0.8% gels which are suitable for separation for the DNA molecule in the range of 0.5 kb ---10 kb. The molecular weight of the DNA can be known from the calibration curve prepared from the known standard of the DNA. The smaller fragment of the DNA moves faster in the gel in compare to the larger fragments. Also open DNA moves slower in compare to the closed circular DNA. Preparation of the gel Gel is prepared by dissolving agarose in the water according to the need. The volume is calculated by measuring the length and breadth of the gel tank. The gel is boiled and poured in the tank and left for the solidifying. In the mean time, the comb is introduced in the gel, so that well can form in the gel. In this well, DNA material is loaded. The gel is sealed surrounding the gel tank which is later on opened before placing them in the electrophoresis tank. Electrophoresis tank is filled with the buffer. Along with the sample some dye and glycerol is also loaded so that they cant float in the buffer. Bromophenol blue is generally used and also ethidium bromide. Ethidium bromide is introduced in between the DNA double stranded DNA. Polyacrylamide gel Polymerization of the acrylamide results in the formation of the polyacrylamide. So they are also called as PAGE. This requires the presence of smaller amount of the N, N methylene bis acrylamide (bis-acrylamide). Actually the bis acrylamide is two acrylamide joined by the methylene. Polyacrylamide polymerization is the free radical catalysis, and is initiated by the addition of ammonium persulphate and the base N,N,N,N- TETRAMETHYLENE DIAMINE (TEMED). TEMED catalyses the decomposition of the persulphate ion to give a free radical (a molecule with unpaired electron). It must be noted that all oxygen should be removed prior to use of the gel by degassing it in the vacuum. Induction of the polymerization can be done by a method called as photo-polymerization, but it needs riboflavin which generated the free radical. Acrylamide gel can be made with a content between 3 to 30 %. In SDS gel, 10% to 20 % acrylamide are used in techniques such as SDS gel electrophoresis.
Chapter -13 Enzyme Immobilization Introduction The word "immobilized enzyme" was coined by Katchalski-Katzir in 1971 (KatchalskiKatzir, 1993). Enzymes may be immobilized on solid carriers by various techniques, such as carrier-binding, cross-linking, or entrapment (Katchalski-Katzir, 1993). Immobilization requires carriers to bind and to support the enzymes. A variety of carriers have been tested with different enzymes. Some of these include alginate, magnetite, kappa-carrageenan, polyurethane, etc. There is sufficient variety to accommodate almost all available industrial enzymes. Interestingly, there are very few detailed studies to compare the efficacy of various immobilization methods or immobilization supports. It is generally believed that the best support for one enzyme may not work as well for another enzyme, and that each enzyme ought to be evaluated to determine the optimal carrier matrix system and conditions. Generally, the decision will depend on (a) the various characteristics of the enzymes; (b) requirements of the specific application, ie. the operational conditions; and (c) the properties and limitations of the support system (Bickerstaff, 1997). Aim of Enzyme Immobilization Enzyme immobilization is aimed to restrict the freedom of movement of an enzyme. When one wants to do enzyme immobilization, one must select the carrier (support, matrix) and decide on the method of immobilization. Carriers or support may be selected on the basis of the following considerations:
[a] Physical properties Strength, available surface area, shape or form (eg. Beads, sheets, fibers), compressibility of carriers, porosity, pore volume, permeability, density, flow rate, pressure drop are the main requirements of the immobilization. [b] Chemical properties chemical property like hydrophilicity, inertness to enzyme(s), substrate(s) or cofactor(s), available functional groups for modification, ability to be regenerated or reused. Compatibility with certain buffers e.g. Alginate is not compatible with phosphate buffer. [c] Stability Immobilized enzyme must have stability on storage, residual enzyme activity on storage, mechanical stability of support material when subject to pressure or water flow. [d] Resistance Immobilized enzyme must be resistance against bacterial or fungal attack, disruption by chemicals, pH, temperature, organic solvents, and enzymes such as proteases. [e] Safety Immobilized enzyme must be safe from toxicity of component reagents, health and safety for process workers and end product users. Carriers must be "safe" if the end product is to be used for food, medical or pharmaceutical applications. For example, acrylamide is toxic. [f] Economic Availability and cost of carrier materials, chemicals, special equipment, reagents and technical skills is required. Beside this industrial scale chemical preparation, feasibility for scale-up, continuous processing, effective working life, and re-usability must not be very costly. Reaction: Immobilized enzymes must not show any diffusion limitations on mass transfer of cofactors, substrates or products, Side reactions. Immobilized enzymes must not be biodegradability for example in waste treatment polyurethane is not naturally occurring and is not easily degraded. Therefore, its use may be undesirable for cleanup. Limitations of Immobilized Enzyme 1. An important factor determining the use of enzymes in a technological process is their expense. Several hundred enzymes are commercially available that are very costly, although some are much cheaper and many are much more expensive. As enzymes are catalytic molecules, they are not directly used up by the processes in which they are used. Their high initial cost, therefore, should only be incidental to their use. 2. However due to denaturation, they do lose activity with time. If possible, they should be stabilised against denaturation and utilised in an efficient manner. 3. When they are used in a soluble form, they retain some activity after the reaction which cannot be economically recovered for re-use and is generally wasted. This activity residue remains to contaminate the product and its removal may involve extra purification costs. In order to eliminate this wastage, and give an improved productivity, simple and economic methods must be used which enable the separation of the enzyme from the reaction product. The easiest way of achieving this is by separating the enzyme and product during the reaction using a two-phase system; one phase containing the enzyme and the other phase containing the product. The enzyme is imprisoned within its phase allowing its re-use or continuous use but preventing it from contaminating the product; other molecules, including the reactants, are able to move freely between the two phases. This is known as immobilisation and may be achieved by fixing the enzyme to, or within, some other material. The term 'immobilisation' does not necessarily mean that the enzyme cannot move freely within its particular phase, although this is often the case. A wide variety of insoluble materials, also known as substrates (not to be confused with the enzymes' reactants), may be used to immobilise the enzymes by making them insoluble. These are usually inert polymeric or inorganic matrices.
Immobilisation of enzymes often incurs an additional expense and is only undertaken if there is a sound economic or process advantage in the use of the immobilised, rather than free (soluble), enzymes. Advantages of Immobilizations 1. The most important benefit derived from immobilisation is the easy separation of the enzyme from the products of the catalysed reaction. This prevents the enzyme contaminating the product, minimising downstream processing costs and possible effluent handling problems, particularly if the enzyme is noticeably toxic or antigenic. 2. It also allows continuous processes to be practicable, with a considerable saving in enzyme, labour and overhead costs. Immobilisation often affects the stability and activity of the enzyme, but conditions are usually available where these properties are little changed or even enhanced. 3. The productivity of an enzyme, so immobilised, is greatly increased as it may be more fully used at higher substrate concentrations for longer periods than the free enzyme. Insoluble immobilised enzymes are of little use, however, where any of the reactants are also insoluble, due to steric difficulties. Methods of immobilizations There are four principal methods available for immobilising enzymes (Figure 3.1): I. adsorption II. covalent binding III. entrapment IV. membrane confinement Figure 6.1. Immobilised enzyme systems. (a) enzyme non-covalently adsorbed to an insoluble particle; (b) enzyme covalently attached to an insoluble particle; (c) enzyme entrapped within an insoluble particle by a cross-linked polymer; (d) enzyme confined within a semipermeable membrane. ________________________________________ Carrier matrices Carrier matrices for enzyme immobilisation by adsorption and covalent binding must be chosen with care. Of particular relevance to their use in industrial processes is their cost relative to the overall process costs; ideally they should be cheap enough to discard. The manufacture of high-valued products on a small scale may allow the use of relatively expensive supports and immobilisation techniques whereas these would not be economical in the large-scale production of low added-value materials. A substantial saving in costs occurs where the carrier may be regenerated after the useful lifetime of the immobilised enzyme. The surface density of binding sites together with the volumetric surface area sterically available to the enzyme, determine the maximum binding capacity. The actual capacity will be affected by the number of potential coupling sites in the enzyme molecules and the electrostatic charge distribution and surface polarity (i.e. the hydrophobic-hydrophilic balance) on both the enzyme and support. The nature of the support will also have a considerable affect on an enzyme's expressed activity and apparent kinetics. The form, shape, density, porosity, pore size distribution, operational stability and particle size distribution of the supporting matrix will influence the reactor configuration in which the immobilised biocatalyst may be used. The ideal support is cheap, inert, physically strong and stable. It will increase the enzyme specificity (kcat/Km) whilst reducing product inhibition, shift the pH optimum to the desired value for the process, and discourage microbial growth and non-specific adsorption. Some matrices possess other properties which are useful for particular purposes such as ferromagnetism (e.g. magnetic iron oxide, enabling transfer of the biocatalyst by means of magnetic fields), a catalytic surface (e.g. manganese dioxide, which catalytically removes the inactivating hydrogen peroxide produced by most oxidases), or a reductive surface environment (e.g. titania, for enzymes inactivated by oxidation). Clearly most supports possess only some of these
features, but a thorough understanding of the properties of immobilised enzymes does allow suitable engineering of the system to approach these optimal qualities. Adsorption of enzymes In adsorption, the enzyme is bound to the carrier material via reversible surface interactions. The forces involved are electrostatic, such as van der Waals forces, ionic and H-bonding interaction, and possibly hydrophobic forces. The forces are generally weak, but there are sufficiently large to allow reasonable binding. The carriers with adsorption properties are selected on the basis of knowledge of their compatibility with the enzyme. Adsorption utilizes existing surface interactions between enzyme and carrier, and does not require chemical activation or modification. Normally, one does not see any damage to the enzyme by the carriers. Figure 6.2 showing the carrier that adsorbs the Enzyme Some advantages of adsorption include 1. Little or no damage to the enzyme. 2. Simple, cheap and immobilization can be done quickly. 3. No chemical changes to carriers or enzyme. 4. Easily reversible. Some disadvantages of adsorption include Desorption or leakage of enzyme from support. Desorption may occur on changing environmental conditions (such as pH, temperature, ionic strength) or on conformational changes arising from substrate/cofactor binding, contaminant binding, etc. Some physical factors such as flow rate, agitation by stirring, collision or abrasion can cause desorption. Non-specific binding by substrate, cofactor or contaminants to the carrier may result in diffusion limitations and mass transfer problems. This may in turn change the kinetic properties of the reactions. Binding of protons to the support may change the pH of the microenvironment and change the reaction rate. Overloading of carrier possible. This may result in reduced catalytic activity. Possible steric hindrance by the carrier material. Adsorption of enzymes on to insoluble supports is a very simple method of wide applicability and capable of high enzyme loading (about one gram per gram of matrix). Simply mixing the enzyme with a suitable adsorbent, under appropriate conditions of pH and ionic strength, followed, after a sufficient incubation period, by washing off loosely bound and unbound enzyme will produce the immobilised enzyme in a directly usable form (Figure 3.2). The driving force causing this binding is usually due to a combination of hydrophobic effects and the formation of several salt links per enzyme molecule. The particular choice of adsorbent depends principally upon minimising leakage of the enzyme during use. Although the physical links between the enzyme molecules and the support are often very strong, they may be reduced by many factors including the introduction of the substrate. Care must be taken that the binding forces are not weakened during use by inappropriate changes in pH or ionic strength. Examples of suitable adsorbents are ion-exchange matrices (Table 3.1), porous carbon, clays, hydrous metal oxides, glasses and polymeric aromatic resins. Ion-exchange matrices, although more expensive than these other supports, may be used economically due to the ease with which they may be regenerated when their bound enzyme has come to the end of its active life; a process which may simply involve washing off the used enzyme with concentrated salt solutions and re-suspending the ion exchanger in a solution of active enzyme. ________________________________________ Figure 6.2. Schematic diagram showing the effect of soluble enzyme concentration on the activity of enzyme immobilised by adsorption to a suitable matrix. The amount adsorbed depends on the incubation time, pH, ionic strength, surface area, porosity, and the physical characteristics of both the enzyme and the support.
________________________________________ Table 6.1 Preparation of immobilised invertase by adsorption (Woodward 1985) Support type % bound at DEAE-Sephadex anion exchanger CM-Sephadex cation exchanger pH 2.5 0 100 pH 4.7 100 75 pH 7.0 100 34 ________________________________________ Covalent coupling This involves the formation of a covalent bond between the enzyme and the carrier material. The bond is normally formed between functional groups on the carrier and the enzyme. Those on the enzymes are usually amino acid residues such as amino (NH2) group from Lys or Arg, carboxyl (COOH) group from Asp, Glu, hydroxyl (OH) group from Ser, Thr, and sulfhydryl (SH) group from Cys. Tests must be run to ensure the formation of covalent bonds will not inactivate the enzyme. Through chemical modifications, functional groups on the carriers can be altered to different forms to accommodate different kinds of covalent bonds to be formed with the enzyme. For example, chemical modification of -OH group can give rise to AEcellulose (aminoethyl), CM- cellulose (carboxymethyl), and DEAE-cellulose (diethylaminoethyl). This increases the range of immobilization methods that can be used for a given carrier. While many supports exist, an important factor for enzyme immobilization appears to be hydrophilicity, which helps to maintain enzyme activity in a hydrophilic milieu. Immobilisation of enzymes by their covalent coupling to insoluble matrices is an extensively researched technique. Only small amounts of enzymes may be immobilised by this method (about 0.02 gram per gram of matrix) although in exceptional cases as much as 0.3 gram per gram of matrix has been reported. The strength of binding is very strong, however, and very little leakage of enzyme from the support occurs. Functional groups that affects the covalent coupling The relative usefulness of various groups, found in enzymes, for covalent link formation depends upon their availability and reactivity (nucleophilicity), in addition to the stability of the covalent link, once formed (Table 3.2). The reactivity of the protein side-chain nucleophiles is determined by their state of protonation (i.e. charged status) and roughly follows the relationship -S- > -SH > -O- > -NH2 > -COO- > -OH >> -NH3+where the charges may be estimated from a knowledge of the pKa values of the ionising groups (Table 1.1) and the pH of the solution. Lysine residues are found to be the most generally useful groups for covalent bonding of enzymes to insoluble supports due to their widespread surface exposure and high reactivity, especially in slightly alkaline solutions. They also appear to be only very rarely involved in the active sites of enzymes. ________________________________________ Table 6.2 Relative usefulness of enzyme residues for covalent coupling Residue Content Exposure Reactivity Stability of couple Use Aspartate + ++ + + + Arginine + ++ Cysteine ++ Cystine + Glutamate + ++ + + + Histidine ++ + + + Lysine ++ ++ ++ ++ ++ Methionine Serine ++ + + Threonine ++ + Tryptophan -
Tyrosine + + C terminus ++ N terminus ++ Carbohydrate - ~ ++ Others - ~ ++ -
_+ + ++ ++ -
+ + + ++ + + + - ~ ++
The most commonly used method for immobilising enzymes on the research scale (i.e. using less than a gram of enzyme) involves Sepharose, activated by cyanogen bromide. This is a simple, mild and often successful method of wide applicability. Sepharose is a commercially available beaded polymer which is highly hydrophilic and generally inert to microbiological attack. Chemically it is an agarose (poly{1,3-D-galactose--1,4-(3,6-anhydro)-L-galactose}) gel. The hydroxyl groups of this polysaccharide combine with cyanogen bromide to give the reactive cyclic imido-carbonate. This reacts with primary amino groups (i.e. mainly lysine residues) on the enzyme under mildly basic conditions (pH 9 - 11.5, Figure 3.3a). The high toxicity of cyanogen bromide has led to the commercial, if rather expensive, production of ready-activated Sepharose and the investigation of alternative methods, often involving chloroformates, to produce similar intermediates (Figure 3.3b). Carbodiimides (Figure 13.3c) are very useful bifunctional reagents as they allow the coupling of amines to carboxylic acids. Careful control of the reaction conditions and choice of carbodiimide allow a great degree of selectivity in this reaction. Glutaraldehyde is another bifunctional reagent which may be used to cross-link enzymes or link them to supports (Figure 13.3d). It is particularly useful for producing immobilised enzyme membranes, for use in biosensors, by cross-linking the enzyme plus a non-catalytic diluent protein within a porous sheet (e.g. lens tissue paper or nylon net fabric). The use of trialkoxysilanes allows even such apparently inert materials as glass to be coupled to enzymes (Figure 13.3e). There are numerous other methods available for the covalent attachment of enzymes (e.g. the attachment of tyrosine groups through diazo-linkages, and lysine groups through amide formation with acyl chlorides or anhydrides). ________________________________________ A. cyanogen bromide [6.3] . (a) Activation of Sepharose by cyanogen bromide. Conditions are chosen to minimise the formation of the inert carbamate. (b) Ethyl chloroformate [6.3] (b) Chloroformates may be used to produce similar intermediates to those produced by cyanogen bromide but without its inherent toxicity. (c) Carbodiimide [6.3] (c) Carbodiimides may be used to attach amino groups on the enzyme to carboxylate groups on the support or carboxylate groups on the enzyme to amino groups on the support. Conditions are chosen to minimise the formation of the inert substituted urea. (d) Glutaraldehyde [6.3] (d) Glutaraldehyde is used to cross-link enzymes or link them to supports. It usually consists of an equilibrium mixture of monomer and oligomers. The product of the condensation of enzyme and glutaraldehyde may be stabilised against dissociation by reduction with sodium borohydride. (e) 3-aminopropyltriethoxysilane Figure 6.3. (e) The use of trialkoxysilane to derivatise glass. The reactive glass may be linked to enzymes by a number of methods including the use thiophosgene, as shown. ________________________________________
It is clearly important that the immobilised enzyme retains as much catalytic activity as possible after reaction. This can, in part, be ensured by reducing the amount of enzyme bound in non-catalytic conformations (Figure 3.4). Immobilisation of the enzyme in the presence of saturating concentrations of substrate, product or a competitive inhibitor ensures that the active site remains unreacted during the covalent coupling and reduces the occurrence of binding in unproductive conformations. The activity of the immobilised enzyme is then simply restored by washing the immobilised enzyme to remove these molecules. ________________________________________ Figure 6.4. The effect of covalent coupling, on the expressed activity of an immobilised enzyme,. (a) Immobilised enzyme, (E) with its active site unchanged and ready to accept the substrate molecule (S), as shown in (b). (c) Enzyme bound in a non-productive mode due to the inaccessibility of the active site. (d) Distortion of the active site produces an inactive immobilised enzyme. Nonproductive modes are best prevented by the use of large molecules reversibly bound in or near the active site. Distortion can be prevented by use of molecules which can sit in the active site during the coupling process, or by the use of a freely reversible method for the coupling which encourages binding to the most energetically stable (i.e. native) form of the enzyme. Both (c) and (d) may be reduced by use of 'spacer' groups between the enzyme and support, effectively displacing the enzyme away from the steric influence of the surface. ________________________________________ Entrapment and Encapsulation In both of these methods, the enzyme remains free in solution, but restricted in movement by the lattice structure of a gel. The porosity of the gel lattice is controlled to ensure that the structure is tight enough to prevent leakage of enzyme, while allowing free movement of substrates, cofactors and products. Encapsulation generally refers to larger capsules that allow for co-immobilization of different combinations of enzymes for selected applications. Notably, red blood cells can be used as encapsulation capsules. Entrapment of enzymes Entrapment of enzymes within gels or fibers is a convenient method for use in processes involving low molecular weight substrates and products. Amounts in excess of 1 g of enzyme per gram of gel or fiber may be entrapped. However, the difficulty which large molecules have in approaching the catalytic sites of entrapped enzymes precludes the use of entrapped enzymes with high molecular weight substrates. The entrapment process may be a purely physical caging or involve covalent binding. As an example of this latter method, the enzymes' surface lysine residues may be derivitized by reaction with acryloyl chloride (CH2=CH-CO-Cl) to give the acryloyl amides. This product may then be copolymerised and cross-linked with acrylamide (CH2=CH-CO-NH2) and bisacrylamide (H2N-CO-CH=CHCH=CH-CO-NH2) to form a gel. Enzymes may be entrapped in cellulose acetate fibers by, for example, making up an emulsion of the enzyme plus cellulose acetate in methylene chloride, followed by extrusion through a spinneret into a solution of an aqueous precipitant. Entrapment is the method of choice for the immobilisation of microbial, animal and plant cells, where calcium alginate is widely used. Membrane confinement Membrane confinement of enzymes may be achieved by a number of quite different methods, all of which depend for their utility on the semipermeable nature of the membrane. This must confine the enzyme whilst allowing free passage for the reaction products and, in most configurations, the substrates. The simplest of these methods is achieved by placing the enzyme on one side of the semipermeable membrane whilst the reactant and product stream is present on the other side. Hollow fiber membrane units are available commercially with large surface areas relative to their contained volumes (> 20 m2 l-1) and permeable only to substances of molecular weight substantially less than the enzymes. Although costly, these are very easy to use for a wide variety of enzymes (including regenerating
coenzyme systems, see Chapter 8) without the additional research and development costs associated with other immobilisation methods. Encapsulation of Enzymes Enzymes, encapsulated within small membrane-bound droplets or liposomes, may also be used within such reactors. As an example of the former, the enzyme is dissolved in an aqueous solution of 1,6-diaminohexane. This is then dispersed in a solution of hexanedioic acid in the immiscible solvent, chloroform. The resultant reaction forms a thin polymeric (Nylon-6,6) shell around the aqueous droplets which traps the enzyme. Liposomes are concentric spheres of lipid membranes, surrounding the soluble enzyme. They are formed by the addition of phospholipids to enzyme solutions. The micro-capsules and liposomes are washed free of non-confined enzyme and transferred back to aqueous solution before use. Table 6.3 presents a comparison of the more important general characteristics of these methods. ________________________________________ Table 6.3 Generalised comparison of different enzyme immobilisation techniques. Characteristics Adsorption Covalent binding Entrapment Membrane confinement Preparation Simple Difficult Difficult Simple Cost Low High Moderate High Binding force Variable Strong Weak Strong Enzyme leakage Yes No Yes No Applicability Wide Selective Wide Very wide Running Problems High Low High High Matrix effects Yes Yes Yes No Large diffusional barriers No No Yes Yes Microbial protection No No Yes Yes ________________________________________ ________________________________________ There are several methods of immobilization (Bickerstaff, 1997), as listed below. Methods compatible with the enzyme, substrate or cofactor should be selected. For example, the entrapment method may not work well with cellulose substrate because the substrate itself is large and will not readily go into the entrapment matrix to reach the enzyme. Crosslinking This method joins the enzyme to each other to form a large, 3-D structure without any support. This can be done through chemical (covalent bonding) or physical means (flocculation). Tests must be done to ensure the active site remains free and available for catalytic activity. Encapsulation often improves the operational stability and sometimes efficiency of enzyme catalysis under industrial conditions, and also allows their easy separation from the medium. This allows for easier recovery of the enzymes for repeat use. The improved stability and ease of separation of immobilized enzymes make them suitable to be used in a variety of bioreactors. They are also easier to scale up. In addition, encapsulation allows for ease of handling and storage of enzymes. Sometimes other properties of the enzyme may be altered by immobilization, e.g. altered pH-activity profile.
Chapter-14 Introduction to Industrial biotechnology Industrial biotechnology applies the techniques of modern molecular biology to improve the efficiency and reduce the environmental impacts of processes in industries like food production, grain cleaning, textiles, paper and pulp and specialty chemicals. Just as biotechnology is transforming the pharmaceutical industry, some observers predict the same impact in the industrial sector. Today
up to 90% of the enzymes used in large scale, for commercial applications result from the exploitation of rDNA methods in the manufacturing process or for the improvement of the catalysts themselves. The progress made in applying the techniques of genetic engineering in the development of industrial-scale processes that produce or utilize enzymes has been simply amazing. Both improved economics as well as the manufacture of novel products not possible or practical by traditional chemical approaches have been achieved. Even today, much of the science is focused on the development of new technological approaches that will allow the future solution of problems of fundamental understanding as well as practical application. Industrial biotechnology companies develop biocatalysts, such as enzymes, to be used in chemical synthesis. Enzymes are proteins produced by all living organisms. In humans, enzymes help digest food, signal cells to turn on and off and perform other complex functions. Other animals use enzymes to break cellulose into sugar or break up proteins. Scientists can locate enzymes in the natural environment with special functional characteristics that have significant commercial value. Using biotechnology, the desired enzyme can then be manufactured in commercial quantities. Most enzymes are manufactured in fermentation systems like human therapeutic proteins. The manufacturing process uses renewable resources as a raw material feedstock. Contrary to inorganic catalysts such as acids, bases, metals and metal oxides, enzymes are very specific. In other words, each enzyme can break down or synthesize one particular compound. In some cases, they limit their action to specific bonds in the compounds with which they react. Most proteases, for instance, can break down several types of protein, but in each protein molecule only certain bonds will be cleaved depending on which enzyme is used. In industrial processes, the specific action of enzymes allows high yields to be obtained with a minimum of unwanted byproducts. Enzymes are very efficient catalysts. For example, the enzyme catalase, which is found abundantly in the liver and red blood cells, is so efficient that in one minute one enzyme molecule can catalyze the breakdown of five million molecules of hydrogen peroxide into water and oxygen.Being formed to work in living cells, enzymes can work at atmospheric pressure and in mild conditions in terms of temperature and acidity. Most enzymes function optimally at a temperature of 30-70C and at pH values which are near pH 7. For certain technical applications, special enzymes have been developed that work at higher temperatures. However, no enzyme can withstand temperatures above 100C for long. Enzyme processes are therefore potentially energysaving and save investing in special equipment resistant to heat, pressure or corrosion. Due to their efficiency, specific action, the mild conditions in which they work and their high biodegradability, enzymes are very well suited to a wide range of industrial applications.Nature provides rich resources for innovative solutions for many industries. Companies involved in industrial biotechnology are constantly striving to discover and develop high-value enzymes and bioactive compounds that will enhance current industrial processes. Chemical processes, including paper manufacturing, textile processing, oil field chemicals and specialty chemical synthesis reactions, sometimes need to run at very high or very low temperatures or high or low pHs. The challenge is to find organisms that can survive and even thrive in these environments. The opportunity to identify unique bioactive molecules is vast. Less than 1 percent of the microorganisms in the world have been cultured and characterized. Application of Immobilised-Enzyme Processes Introduction Immobilise -enzyme systems are used where they offer cost advantages to users on the basis of total manufacturing costs. The plant size needed for continuous processes is two orders of magnitude smaller than that required for batch processes using free enzymes. The capital costs are, therefore, considerably smaller and the plant may be prefabricated cheaply off-site Immobilised enzymes
offer greatly increased productivity on an enzyme weight basis and also often provide process advantages (see Chapter 6) Currently used immobilised-enzyme processes are given in Table 7.1. ________________________________________ Table 7.1 Some of the more important industrial uses of immobilised enzymes Enzyme EC number Product Aminoacylase 3.5.1.14 L-Amino acids Aspartate ammonia-lyase 4.3.1.1 L-Aspartic acid Aspartate 4-decarboxylase 4.1.1.12 L-Alanine Cyanidase 3.5.5.x Formic acid (from waste cyanide) Glucoamylase 3.2.1.3 D-Glucose Glucose isomerase 5.3.1.5 High -fructose corn syrup Histidine ammonia-lyase 4.3.1.3 Urocanic acid Hydantoinasea 3.5.2.2 D- and L-amino acids Invertase 3.2.1.26 Invert sugar Lactase 3.2.1.23 Lactose-free milk and whey Lipase 3.1.1.3 Cocoa butter substitutes Nitrile hydratase 4.2.I.x Acrylamide Penicillin amidases 3.5.1.11 Penicillins Raffinase 3.2.1.22 Raffinose-free solutions Thermolysin 3.2.24.4 Aspartame a Dihydropyrimidinase 1-High-fructose corn syrups (HFCS) With the development of glucoamylase in the 1940s and 1950s it became a straightforward matter to produce high DE glucose syrups. However, these have shortcomings as objects of commerce: D-glucose has only about 70% of the sweetness of sucrose, on a weight basis, and is comparatively insoluble. Batches of 97 DE glucose syrup at the final commercial concentration (71% (w/w)) must be kept warm to prevent crystallisation or diluted to concentrations that are microbiologically insecure. Fructose is 30% sweeter than sucrose, on a weight basis, and twice as soluble as glucose at low temperatures so a 50% conversion of glucose to fructose overcomes both problems giving a stable syrup that is as sweet as a sucrose solution of the same concentration (see Table 14.3). The isomerisation is possible by chemical means but not economical, giving tiny yields and many by-products (e.g. 0.1 M glucose 'isomerised' with 1.22 M KOH at 5C under nitrogen for 3.5 months gives a 5% yield of fructose but only 7% of the glucose remains unchanged, the majority being converted to various hydroxy acids). 2-GLUCOSE ISOMERASE Glucose is normally isomerised to fructose during glycolysis but both sugars are phosphorylated. The use of this phosphohexose isomerase may be ruled out as a commercial enzyme because of the cost of the ATP needed to activate the glucose and because two other enzymes (hexokinase and fructose-6-phosphatase) would be needed to complete the conversion. Only an isomerase that would use underivatised glucose as its substrate would be commercially useful but, until the late 1950s, the existence of such an enzyme was not suspected. At about this time, enzymes were found that catalyse the conversion of D-xylose to an equilibrium mixture of D-xylulose and D-xylose in bacteria. When supplied with cobalt ions, these xylose isomerases were found to isomerise -D-glucopyranose to -D fructofuranose. Now it is known that several genera of microbes, mainly bacteria, can produce such glucose isomerases: The commercial enzymes are produced by Actinoplanes missouriensis, Bacillus coagulans and various Streptomyces species; as they have specificities for glucose and fructose which are not much different from that for xylose and ways are being found to avoid the necessity of xylose as inducer, these should perhaps now no longer be considered as xylose isomerases. They are remarkably friendly enzymes in that they are resistant to thermal denaturation and will act at very high substrate concentrations, which have the additional benefit of substantially stabilising the
enzymes at higher operational temperatures. The vast majority of glucose isomerases are retained within the cells that produce them but need not be separated and purified before use. All glucose isomerases are used in immobilised forms. Although differerent immobilisation methods have been used for enzymes from differerent organisms, the principles of use are very similar. Immobilisation is generally by cross-linking with glutaraldehyde, plus in some cases a protein diluent, after cell lysis or homogenisation. Originally, immobilised glucose isomerase was used in a batch process. This proved to be costly as the relative reactivity of fructose during the long residence times gave rise to significant by -product production. Also, difficulties were encountered in the removal of the added Mg2+ and Co2+ and the recovery of the catalyst. Nowadays most isomerisation is performed in PBRs (Table 14.2). They are used with high substrate concentration (35-45% dry solids, 93-97% glucose) at 5560C. The pH is adjusted to 7.5-8.0 using sodium carbonate and magnesium sulphate is added to maintain enzyme activity (Mg2+ and Co2+ are cofactors). The Ca2+ concentration of the glucose feedstock is usually about 25 m, left from previous processing, and this presents a problem. Ca2+ competes successfully for the Mg2+ binding site on the enzyme, causing inhibition. At this level the substrate stream is normally made 3 mM with respect to Mg2+. At higher concentrations of calcium a Mg2+ : Ca2+ ratio of 12 is recommended. Excess Mg2+ is uneconomic as it adds to the purification as well as the isomerisation costs. The need for Co2+ has not been eliminated altogether, but the immobilisation methods now used fix the cobalt ions so that none needs to be added to the substrate streams. ________________________________________ Table 7.2. Comparison of glucose isomerisation methods Parameter Batch (soluble GI) Batch (immobilised Gl) Continuous (PBR) Reactor volume (m3) 1100 1100 15 Enzyme consumption (tonnes) 180 11 2 Activity, half-life (h) 30 300 1500 Active life, half-lives 0.7 2 3 Residence time (h) 20 20 0.5 Co2+ (tonnes) 2 1 0 Mg2+ (tonnes) 40 40 7 Temperature (C) 65 65 60 pH 6.8 6.8 7.6 Colour formation (A420) 0.7 0.2 < 0.1 Product refining Filtration C-treatmenta Cation exchange Anion exchange C-treatment Cation exchange Anion exchange C -treatment Capital, labour and energy costs, tonne-1 5 5 1 Conversion cost, tonne-1 500 30 5 All processes start with 45% (w/w) glucose syrup DE 97 and produce 10000 tonnes per month of 42% fructose dry syrup. Some of the improvement that may be seen for PBR productivity is due to the substantial development of this process. a Treatment with activated carbon. ________________________________________
Precautions It is essential for efficient use of immobilised glucose isomerase that the substrate solution is adequately purified so that it is free of insoluble material and other impurities that might inactivate the enzyme by chemical (inhibitory) or physical (pore-blocking) means. In effect, this means that glucose produced by acid hydrolysis cannot be used, as its low quality necessitates extensive and costly purification. Insoluble material is removed by filtration, sometimes after treatment with flocculants, and soluble materials are removed by ion exchange resins and activated carbon beads. This done, there still remains the possibility of inhibition due to oxidised by-products caused by molecular oxygen. This may be removed by vacuum de-aeration of the substrate at the isomerisation temperature or by the addition of low concentrations (< 50 ppm) of sulphite. At equilibrium at 60C about 51 % of the glucose in the reaction mixture is converted to fructose. However, because of the excessive time taken for equilibrium to be attained and the presence of oligosaccharides in the substrate stream, most manufacturers adjust flow rates so as to produce 42-46% (w/w) fructose (leaving 47-51 % (w/w) glucose). To produce 100 tonnes (dry substance) of 42% HFCS per day, an enzyme bed volume of about 4 m3 is needed. Activity decreases, following a first-order decay equation. The half-life of most enzyme preparations is between 50 and 100 days at 55C. Typically a batch of enzyme is discarded when the activity has fallen to an eighth of the initial value (i.e. after three half -lives). To maintain a constant fructose content in the product, the feed flow rate is adjusted according to the enzyme activity. Several reactors containing enzyme preparations of different ages are needed to maintain overall uniform production by the plant (Figure 14.10). In its lifetime 1 kg of immobilised glucose isomerase (exemplified by Novo's Sweetzyme T) will produce 10 -11 tonnes of 42% fructose syrup (dry substance). ________________________________________ Figure 7.10. Diagram showing the production rate of a seven-column PBR facility on start -up, assuming exponential decay of reactor activity. The columns are brought into use one at a time. At any time a maximum of six PBRs are operating in parallel, whilst the seventh, exhausted, reactor is being refilled with fresh biocatalyst. PBR activities allowed to decay through three half-lives (to 12.5% initial activity) before replacement. The final average productivity is 2.51 times the initial productivity of one column. - - -- - - - PBR activities allowed to decay through two half-lives (to 25% initial activity) before replacement. The final average productivity is 3.23 times the initial productivity of one column. It may be seen that the final average production rate is higher when the PBRs are individually operated for shorter periods but this 29% increase in productivity is achieved at a cost of 50% more enzyme, due to the more rapid replacement of the biocatalyst in the PBRs. A shorter PBR operating time also results in a briefer start-up period and a more uniform productivity. ________________________________________ After isomerisation, the pH of the syrup is lowered to 4 - 5 and it is purified by ion-exchange chromatography and treatment with activated carbon. Then, it is normally concentrated by evaporation to about 70% dry solids.For many purposes a 42% fructose syrup is perfectly satisfactory for use but it does not match the exacting criteria of the quality soft drink manufacturers as a replacement for sucrose in acidic soft drinks. For use in the better colas, 55% fructose is required. This is produced by using vast chromatographic columns of zeolites or the calcium salts of cation exchange resins to adsorb and separate the fructose from the other components. The fractionation process, although basically very simple, is only economic if run continuously. The fructose stream (90% (w/w) fructose, 9% glucose) is blended with 42% fructose syrups to give the 55% fructose (42% glucose) product required. The glucose-rich 'raffinate' stream may be recycled but if this is done undesirable oligosaccharides build up in the system. Immobilised glucoamylase is used in some plants to hydrolyse oligosaccharides in
the raffinate; here the substrate concentration is comparatively low (around 20% dry solids) so the formation of isomaltose by the enzyme is insignificant.Clearly the need for a second large fructose enrichment plant in addition to the glucose isomerase plant is undesirable and attention is being paid to means of producing 55% fructose syrups using only the enzyme. The thermodynamics of the system favour fructose production at higher temperatures and 55% fructose syrups could be produced directly if the enzyme reactors were operated at around 95C. The use of miscible organic co -solvents may also produce the desired effect. Both these alternatives present a more than considerable challenge to enzyme technology! The present world market for HFCS is over 5 million tonnes of which about 60% is for 55% fructose syrup with most of the remainder for 42% fructose syrup. This market is still expanding and ensures that HFCS production is the major application for immobilised-enzyme technology.The high-fructose syrups can be used to replace sucrose where sucrose is used in solution but they are inadequate to replace crystalline sucrose. Another ambition of the corn syrup industry is to produce sucrose from starch. This can be done using a combination of the enzymes phosphorylase (EC 2.4.1.1), glucose isomerase and sucrose phosphorylase (EC 2.4.1.7), but the thermodynamics do not favour the conversion so means must be found of removing sucrose from the system as soon as it is formed. This will not be easy but is achievable if the commercial pull (i.e. money available) is sufficient: phosphorylase starch (Gn) + orthophosphate starch (Gn-1) + -glucose-1-phosphate [7.1] glucose isomerase glucose fructose [7.2] sucrose phosphorylase -glucose-1 -phosphate + fructose sucrose + orthophosphate [7.3] A further possible approach to producing sucrose from glucose is to supply glucose at high concentrations to microbes whose response to osmotic stress is to accumulate sucrose intracellularly. Provided they are able to release sucrose without hydrolysis when the stress is released, such microbes may be the basis of totally novel processes. 3-Use of immobilised raffinase The development of a raffinase (-D-galactosidase) suitable for commercial use is another triumph of enzyme technology. Plainly, it would be totally unacceptable to use an enzyme preparation containing invertase to remove this material during sucrose production. It has been necessary to find an organism capable of producing an -galactosidase but not an invertase. A mould, Mortierella vinacea var. raffinoseutilizer, fills the requirements. This is grown in a particulate form and the particles harvested, dried and used directly as the immobilised-enzyme preparation. It is stirred with the sugar beet juice in batch stirred tank reactors. When the removal of raffinose is complete, stirring is stopped and the juice pumped off the settled bed of enzyme. Enzyme, lost by physical attrition, is replaced by new enzyme added with the next batch of juice. The galactose released is destroyed in the alkaline conditions of the first stages of juice purification and does not cause any further problems while the sucrose is recovered. This process results in a 3% increase in productivity and a significant reduction in the costs of the disposal of waste molasses. Immobilised raffinase may also be used to remove the raffinose and stachyose from soybean milk. These sugars are responsible for the flatulence that may be caused when soybean milk is used as a milk substitute in special diets. 4-Use of immobilised Invertase Invertase was probably the first enzyme to be used on a large scale in an immobilised form (by Tate & Lyle). In the period 1941 -1946 the acid, previously used in the manufacture of Golden Syrup, was unavailable, so yeast invertase was used instead. Yeast cells were autolysed and the autolysate clarified by adjustment to pH 4.7, followed by filtration through a bed of calcium sulphate and
adsorption into bone char. A layer of the bone char containing invertase was included in the bed of bone char already used for decolourising the syrup. The scale used was large, the bed of invertase-char being 2 ft (60 cm) deep in a bed of char 20 ft (610 cm) deep. The preparation was very stable, the limiting factors being microbial contamination or loss of decolourising power rather than loss of enzymic activity. The process was cost-effective but, not surprisingly, the product did not have the subtlety of flavour of the acid-hydrolysed material and the immobilised enzyme process was abandoned when the acid became available once again. Recently, however, it has been relaunched using BrimacTM, where the invertase -char mix is stabilised by cross-linking and has a half-life of 90 days in use (pH 5.5, 50C). The revival is due, in part, to the success of HFCS as a high-quality low-colour sweetener. It is impossible to produce inverted syrups of equivalent quality by acid hydrolysis. Enzymic inversion avoids the high-colour, high salt-ash, relatively low conversion and batch variability problems of acid hydrolysis. Although free invertase may be used (with residence times of about a day), the use of immobilised enzymes in a PBR (with residence time of about 15 min) makes the process competitive; the cost of 95% inversion (at 50% (w/w)) being no more than the final evaporation costs (to 75% (w/w)). A productivity of 16 tonnes of inverted syrup (dry weight) may be achieved using one litre of the granular enzyme. 5-Production of amino acids Another early application of an immobilised enzyme was the use of the aminoacylase from Aspergillus oryzae to resolve racemic mixtures of amino acids. Figure [7.4] Chemically synthesised racemic N-acyl-DL-amino acids are hydrolysed at pH 8.5 to give the free L-amino acids plus the unhydrolysed N-acyl-D-amino acids. These products are easily separated by differential crystallisation and the N-acyl-D-amino acids racemised chemically (or enzymically) and reprocessed. The enzyme is immobilised by adsorption to anion exchange resins (e.g. DEAE-Sephadex) and has an operational half-life of about 65 days at 50C in PBRs with residence times of about 30 min. The reactors may be re-activated in situ by simply adding more enzyme. The immobilised enzyme has proved a more economical process than the use of free enzyme mainly due to the more efficient use of the substrate and reductions in the cost of enzyme and labour. Novel and natural L-amino acids can be produced by the chemical conversion of aldehydes through DL-amino nitrites to racemic DL-hydantoins (reaction scheme [14.5]) followed by enzymic hydrolysis with hydantoinase and a carbamoylase (reaction scheme [14.6]) at pH 8.5. Both enzymes may be obtained from Arthrobacter species. D -Amino acids are important constituents in antibiotics and insecticides. They may be produced in a manner similar to the L-amino acids but using hydantoinases of differing specificity. The Pseudomonas striata enzyme is specific for Dhydantoins, allowing their specific hydrolysis to D-carbamoyl amino acids which can be converted to the D-amino acids by chemical treatment with nitrous acid. They remaining L-hydantoin may be simply racemised by base and the process repeated. [7.5] [7.6] L -Aspartic acid is widely used in the food and pharmaceutical industries and is needed for the production of the low -calorific sweetener aspartame. It may be produced from fumaric acid by the use of the aspartate ammonia-lyase (aspartase) from Escherichia coli. aspartate ammonia-lyase -OOCCH=CHCOO- + NH4+ -OOCCH2CH(NH3+)COO[7.7] fumaric acid L-aspartic acid A crude immobilised aspartate ammonia-lyase (50000 U g-1) may be prepared by entrapping Escherichia coli cells in a -carageenan gel crosslinked with glutaraldehyde and hexamethylenediamine. The process is operated in a PBR at pH
8.5 using ammonium fumarate as the substrate, with a reported operational halflife of 680 days at 37C. Urocanic acid is a sun-screening agent which may be produced from L-histidine by the histidine ammonia -lyase (histidase) from Achromobacter liquidum . The organism cannot be used directly as it has urocanate hydratase activity, which removes the urocanic acid. However, a brief heat treatment (70C, 30 min) inactivates this unwanted activity but has little effect on the histidine ammonialyase. A crude immobilised-enzyme preparation consisting of heat -treated cells entrapped in a polyacrylamide gel has been used to effect this conversion, showing a half-life of 180 days at 37C. 6- Use of immobilised lactase Lactase is one of relatively few enzymes that have been used both free and immobilised in large-scale processes. The reasons for its utility has been given earlier , but the relatively high cost of the enzyme is an added incentive for its use in an immobilised state.Immobilised lactases are important mainly in the treatment of whey, as the fats and proteins in the milk emulsion tend to coat the biocatalysts. This both reduces their apparent activity and increases the probability of microbial colonisation. Yeast lactase has been immobilised by incorporation into cellulose triacetate fibres during wet spinning, a process developed by Snamprogetti S.p.A. in Italy. The fibres are cut up and used in a batchwise STR process at 5C (Kluyveromyces lactis, pH optimum 6.4 -6.8, 90 U g-1). Fungal lactases have been immobilised on 0.5 mm diameter porous silica (35 nm mean pore diameter) using glutaraldehyde and -aminopropyltriethoxysilane (Asperigillus niger, pH optimum 3.0 -3.5, 500 U g-1; A. oryzae, pH optimum 4.0 -1.5, 400 U g-1). They are used in PBRs. Due to the different pH optima of fungal and yeast lactases, the yeast enzymes are useful at the neutral pH of both milk and sweet whey, whereas fungal enzymes are more useful with acid whey. Immobilised lactases are particularly affected by two inherent short-comings. Product inhibition by galactose and unwanted oligosaccharide formation are both noticeable under the diffusion-controlled conditions usually prevalent. Both problems may be reduced by an increase in the effectiveness factor and a reduction in the degree of hydrolysis or initial lactose concentration, but such conditions also lead to a reduction in the economic return. The control of microbial contamination within the bioreactors is the most critical practical problem in these processes. To some extent, this may be overcome by the use of regular sanitation with basic detergent and a dilute protease solution. .7 Production of antibiotics Benzylpenicillins and phenoxymethylpenicillins (penicillins G and V, respectively) are produced by fermentation and are the basic precursors of a wide range of semisynthetic antibiotics, e.g. ampicillin. The amide link may be hydrolysed conventionally but the conditions necessary for its specific hydrolysis, whilst causing no hydrolysis of the intrinsically more labile but pharmacologically essential -lactam ring, are difficult to attain. Such specific hydrolysis may be simply achieved by use of penicillin amidases (also called penicillin acylases). Different enzyme preparations are generally used for the hydrolysis of the penicillins G and V, pencillin-V-amidase being much more specific than pencillinG-amidase. Penicillin amidase may be obtained from E. coli and has been immobilised on a number of supports including cyanogen bromide-activated Sephadex G200. It represents one of the earliest successful processes involving immobilised enzymes and is generally used in batch or semicontinuous STR processes (40,000 Ukg1penicillin G, 35C, pH 7.8, 2 h) where it may be reused over 100 times. It has also been used in PBRs, where it has an active life of over 100 days, producing about two tonnes of 6-aminopenicillanic acid kg-1of immobolised enzyme. [7.8]
[7.9] The penicillin-G-amidases may be used 'in reverse' to synthesise penicillin and cephalosporin antibiotics by non -equilibrium kinetically controlled reactions. Ampicillin has been produced by the use of penicillin-G-amidase immobilised by adsorption to DEAE -cellulose in a packed bed column: [7.10] Many other potential and proven antibiotics have been synthesised in this manner, using a variety of synthetic -lactams and activated carboxylic acids. Preparation of acrylamide Acrylamide is an important monomer needed for the production of a range of economically useful polymeric materials. It may be produced by the addition of water to acrylonitrile. CH2=CHCN + H2O CH2=CHCONH2 [14.11] This process may be achieved by the use of a reduced copper catalyst (Cu+); however, the yield is poor, unwanted polymerisation or conversion to acrylic acid (CH2=CHCOOH) may occur at the relatively high temperatures involved (80 -140C) and the catalyst is difficult to regenerate. These problems may be overcome by the use of immobilised nitrile hydratase (often erroneously called a nitrilase). The enzyme from Rhodococcus has been used by the Nitto Chemical Industry Co. Ltd, as it contains only very low amidase activity which otherwise would produce unwanted acrylic acid from the acrylamide. Immobilised nitrile hydratase is simply prepared by entrapping the intact cells in a cross-linked 10% (w/v) polyacrylamide/dimethylaminoethylmethacrylate gel and granulating the product. It is used at 10C and pH 8.0-8.5 in a semibatchwise process, keeping the substrate acrylonitrile concentration below 3% (w/v). Using 1% (w/v) immobilised-enzyme concentration (about 50,000 U l-1) the process takes about a day. Product concentrations of up to 20% (w/v) acrylamide have been achieved, containing negligible substrate and less than 0.02% (w/w) acrylic acid. Acrylamide production using this method is about 4000 tonnes per year. The closely related enzymes cyanidase and cyanide hydratase are used to remove cyanide from industrial waste and in the detoxification of feeds and foodstuffs containing amygdalin (see equation [7.12]). HCN + 2H2O HCOO- + NH4+ [7.12] HCN + H2O HCONH2 [7.13]
Chapter-15 Application of Enzyme in clinical diagnosis and Industries Introduction Most important aspects of enzyme activities is the clinical diagnosis. Before 1940 ,only hydrolytic enzyme were used such as lipase, amylase, phosphatase, Trypsin, and pepsin and in diagnosis only 5% enzyme were in the use. Now a day up to 25% of the enzyme is used today for the clinical diagnosis. This is due to increased knowledge in the metabolic pathway and the role of enzyme. Due to enhancing in knowledge of enzyme it is possible to use it for rationale drug design to inhibit specific enzyme. The major application in the clinical field is the determination of the concentration of the substrate secreted in the body fluids with the help of enzymes and comparing it with the normal fluid concentration. With discoveries of monoclonal antibodies different specific detection become easier with the help of different assay system like ELISA. Development of medical applications for enzymes have been at least as extensive as those for industrial applications, reflecting the magnitude of the potential
rewards: for example, pancreatic enzymes have been in use since the nineteenth century for the treatment of digestive disorders. The variety of enzymes and their potential therapeutic applications are considerable. A selection of those enzymes which have realised this potential to become important therapeutic agents is shown in Table 8.1 . At present, the most successful applications are extracellular: purely topical uses, the removal of toxic substances and the treatment of lifethreatening disorders within the blood circulation. Determination of enzyme activities For the determination of enzyme activities for clinical diagnosis, the most important thing is the availability of enzyme in the blood urine or in the tissue. Most of the clinical assay is done with the help of serum. Urine can also be used. Other body fluids that can be used is the body pleura, peritoneum, pericardium, cerebrospinal canal, synovia, stomoach, deudonium, semen , vagina, RBC and WBC. Activities of the enzyme in the serum may increases due to several reason. For example, due to tissue damage, due to increase cell turnover number, cellular proliferation, and neoplasia. In the disease state a tissue become inflamed and swelling occur and it become nacrotic. In that state it may secrete large amount of the enzymes from the dead cells for example release of glutamate dehydrogenase from the dead mitochondrial cells.Note that the stay of the release enzyme is very sort. For example glutamate S transferases has a half lie of 90 minute. To know amount of or fate of serum enzyme labeled DH injected in the rabbit. Then it has been observed that they were rapidly degraded and removed from the blood plasma. There are some tissue specific enzyme that may be detected from them. For example; acid phosphatases in the prostate and acetylcholineestarase in erythrocyte, .with the elctrophoresis technique we can determine many more enzyme in the serum like alkaline phosphatase, amylase , creatine kinase, ceruloplasmine, , glucose-6 phosphate,dehydrogenase and aspartate aminotransferase. Other most studied enzyme in the blood serum is the LDH lactate dehydrogenase, creatine kinase, and alkaline phosphates. In forensic science main enzyme that is used is the adenosine deaminase, adenylate kinase, carbonate dehydrates and phosphatase esterase, phosphoglucomutase amino peptidase, and lactooylglutathione lyse. 1- Clinical Enzymology of liver disease. The most common disease of the liver is the hepatitis, cirrosis, tumors infection. The enzyme most used in the diagnosis is the (ST or SGOT) aspartate amino transferase, ALT or alanine aminotransferase or serum glutamate pyruvate transaminase(SPGT) , alkaline phosphatase, and - glutamyl transerase. Other enzyme is the lactate dehydrogenase, isocitrate dehydrogense 5 nucleotide and glutathione transferase. Ortnithin carbamoyltransferase is almost exclusive to liver. Serum alkaline phosphatase is mainly important in the liver disease. In the case of cirrhosis or hepatitis, aminotransferase are monitored for several months to follow progress of disease. Obstructive jaundice can be determined by the measurement of aminotransferases and alkaline phosphatase. Sometime these enzyme also secreted in the hepatitis so to decide which is the disease better to measure the ratio. The ratio of glutamate dehydrogenase / aminotranserase changes after one week of the viral hepatitis. The ratio is higher in the case of obstructive jaundice than in viral hepatitis. -glutamyltransferase used extensively in the liver specially in the case of alcoholism. 2- In Heart disease The main disease of the heart is the myocardial infarction and, nacrosis of the heart tissue. Enzymes are released during the nacrosis into the plasma. The three enzymes that is assayed are creatine kinase, aspartate aminotransferase, and lactate dehydrogenase. The first enzyme that was used in the diagnosis of the heart disease was the aspartate aminotransferase, but now creatine kinase, lactate dehydrogenase is used mostly. -amylase -amylase is an endoamylase and hydrolyze amylopectine. Its highest concentration
is found in the intestine and salivary gland. In normal person its concentration is detected in low concentration in the urine and the serum but it is increases many fold during the pencreatitis. Pains occur severely in the upper duodenum. During the pancreatic carcinoma it is found in higher concentration in the pancreas. Also this enzyme is detected during the duodenal ulcers. amylase activity also increases during the mumps, when salivary glands become inflamed. Creatine Kinase and fructose bisphosphate aldolase. Creatine kinase predominantly occurs in the skeletal muscle, cardiac muscle and brain. Its concentration (creatine kinase) raised during brain stroke. Alkaline phosphates This enzyme is detected in the obstructive jaundice. It is also useful in the detection of bone disease like ostomalacia, rickets and hyperparathyroidism. This enzyme is normally found in the serum. This enzyme concentration also increases during the pregnancy. Acid phosphatase. This enzyme is found in highest concentration in the prostrate carcinoma. In Treatment of cancer A major potential therapeutic application of enzymes is in the treatment of cancer. Asparaginase has proved to be particularly promising for the treatment of acute lymphocytic leukemia. Its action depends upon the fact that tumour cells are deficient in aspartate-ammonia ligase activity, which restricts their ability to synthesise the normally non-essential amino acid L-asparagine. Therefore, they are forced to extract it from body fluids. The action of the asparaginase does not affect the functioning of normal cells which are able to synthesize enough for their own requirements, but reduce the free exogenous concentration and so induces a state of fatal starvation in the susceptible tumour cells. A 60% incidence of complete remission has been reported in a study of almost 6000 cases of acute lymphocytic leukemia. The enzyme is administered intravenously. It is only effective in reducing asparagine levels within the bloodstream, showing a halflife of about a day (in a dog). This half-life may be increased 20-fold by use of polyethylene glycol-modified asparaginase. Enzyme deficiencies Phenylketonuria is detected by presence of phenylalanine in the urine. Some inborn error of metabolism is also detected are relatively harmless e.g. albinism , alkaptonuria, Table 8.1 showing List of defective enzyme and their diseases Alkaptonuria Phenylketonuria Maple syrup disease Galactosomia uridyltransferasae Glycogen storage Fructosuria Gaucher disease Tay Sachs Wilson disease Acetalasaemia Xerderma pigmentosa homogenistate 1 oxidase phenylalanine 4 monooxygenase oxo acid decorboxylase galactose-1 phosphate glucose 6 phosphate fructokinase glucocerbrosidase -NAcetykll D-hexosaminidase
P-type ATPase catalase DNA binding protein helicases
Enzyme inhibitors and drug design Inhibition of hydroxymethylglutaryl Co-A reductase (HMGCo-A), during hypercholesterolemia. Other competitive inhibitors of HMGCo-A is namely mevinolin, compactin and monacol K which inhibits the enzyme. Inhibition of xanthin oxidase. Gout disease is characterize by uric acid that is end product of purine breakdown in humans derived from breakdown of nucleic acid. The main inhibitor is used allopurinol. Inhibition of HIV protease.The replication of HIV-1 is the cause of AIDS. The HIV1 protease catalyses this protein sequence. HIV protease is an aspartyl protease having a sequence Asp-thr-Gly at its active site. It resemble pepsin, and other retroviral protease. Many inhibitors have been designed that are analogous like rennin. Figure below showing structure of HIV protease. Figure 8. 1 HIV protease Use of enzyme in determination of concentration of metabolite of clinical importance. There are number of enzymatic advantage method estimate the metabolite in presence of many other substances an inhibitor present in serum may completely inhibit enzyme activity. For the estimation of certain metabolite e.g. glucose, and plasma glycerides enzyme methods are now used almost exclusively e.g. serum creatine and non-enzymic method are still being used in clinical chemistry. Blood glucose In the investigation of diabetes there are three enzymatic method that are available; hexokinase couple with glucose 6-phosphate dehydrogenase, glucose oxidase coupled with peroxidase. The second method is glucose oxidase is more commonly used. ________________________________________ Table 8.2 Some important therapeutic enzymes Enzyme EC number Reaction Use Asparaginase 3.5.1.1 L-Asparagine H2O L-aspartate + NH3 Leukaemia
Collagenase 3.4.24.3 Collagen hydrolysis Skin ulcers Glutaminase 3.5.1.2 L-Glutamine H2O L-glutamate + NH3 Leukaemia Hyaluronidasea 3.2.1.35 Hyaluronate hydrolysis Heart attack Lysozyme 3.2.1.17 Bacterial cell wall hydrolysis Antibiotic Rhodanaseb 2.8.1.1 S2O32- + CN- SO32- + SCNCyanide poisoning Ribonuclease 3.1.26.4 RNA hydrolysis Antiviral -Lactamase 3.5.2.6 Penicillin penicilloate Penicillin allergy Streptokinasec 3.4.22.10 Plasminogen plasmin Blood clots Trypsin 3.4.21.4 Protein hydrolysis Inflammation Uricased 1.7.3.3 Urate + O2 allantoin Gout Urokinasee 3.4.21.31 Plasminogen plasmin Blood clots a Hyaluronoglucosaminidase, b thiosulphate sulfurtransferase,c streptococcal cysteine proteinase d urate oxidase,e plasminogen activator ________________________________________ Disadvantages of Using Enzyme as Therapeutic Agents As enzymes are specific biological catalysts, they should make the most desirable therapeutic agents for the treatment of metabolic diseases. Unfortunately a number of factors severely reduce this potential utility: a) They are too large to be distributed simply within the body's cells. This is the major reason why enzymes have not yet been successful applied to the large number of human genetic diseases. A number of methods are being developed in order to overcome this by targeting enzymes; as examples, enzymes with covalently attached external -galactose residues are targeted at hepatocytes and enzymes covalently coupled to target-specific monoclonal antibodies are being used to avoid non-specific side-reactions. b) Being generally foreign proteins to the body, they are antigenic and can elicit an immune response which may cause severe and life-threatening allergic reactions, particularly .on continued use. It has proved possible to circumvent this problem, in some cases, by disguising the enzyme as an apparently nonproteinaceous molecule by covalent modification. Asparaginase, modified by covalent attachment of polyethylene glycol, has been shown to retain its anti-tumour effect whilst possessing no immunogenicity. Clearly the presence of toxins, pyrogens and other harmful materials within a therapeutic enzyme preparation is totally forbidden. Effectively, this encourages the use of animal enzymes, in spite of their high cost, relative to those of microbial origin. Their effective lifetime within the circulation may be only a matter of minutes. This has proved easier than the immunological problem to combat, by disguise using covalent modification. Other methods have also been shown to be successful, particularly those involving entrapment of the enzyme within artificial liposomes, synthetic microspheres and red blood cell ghosts. However, although these methods are efficacious at extending the circulatory lifetime of the enzymes, they often cause increased immunological response and additionally may cause blood clots. In contrast to the industrial use of enzymes, therapeutically useful enzymes are required in relatively tiny amounts but at a very high degree of purity and
(generally) specificity. The favoured kinetic properties of these enzymes are low Km and high Vmax in order to be maximally efficient even at very low enzyme and substrate concentrations. Thus the sources of such enzymes are chosen with care to avoid any possibility of unwanted contamination by incompatible material and to enable ready purification. Therapeutic enzyme preparations are generally offered for sale as lyophilised pure preparations with only biocompatible buffering salts and mannitol diluent added. The costs of such enzymes may be quite high but still comparable to those of competing therapeutic agents or treatments. As an example, urokinase (a serine protease, see Table 4.4) is prepared from human urine (some genetically engineered preparations are being developed) and used to dissolve blood clots. The cost of the enzyme is about Rs. 5000 mg-1, with the cost of treatment in a case of lung embolism being about Rs. 50000 for the enzyme alone. In spite of this, the market for the enzyme is worth about Rs 350M year-1. INDUSTRIAL ENZYME PROCESS AT HIGH TEMPERATURE 8.3 Enzyme operating temperature 0C Major applications alpha amylase (bacterial) 90-100 starch hydroysis, brewing baking detergents Gluco amylase 50-60 Maltodextrin hydrolysis alpha amylase (fungal) 50-60 Maltose Pullulanase 50-60 high glucose syrups Xylose isomerase 45-55 High fructose syrups Pectinase 20-50 clarification of juices /wines Cellulose 45-55 cellulose hydrolysis Lactase 30-50 lactose hydrolysis , food processing Acid protease 30-50 food processing fungal protease 40-60 baking ,brewing,food processing Alkaline proteases 40-60 Detergents Lipases 30-70 Detergents, food processing Enzyme used in Industries Thermozymes Thermozymes are thermostable enzymes that function optimally at 60 0C and 125 0C. these extremozymes are extremely valuable tools for study of protein stability Thermozymes are used in the molecular biology experiments for example taq polymerase and in the detergents (e.g.) proteases and starch processing (e.g. alpha amylase, glucsoe isomerases) various lipase and proteases oxidoreductase are used in the diagnostics, waste treatment and pulp and paper manufacture. Thermozymes are more stable because they are active against denaturing condition Hyperthermophiles have good potential for use in novel biotechnological processes including oil coal and waste gas desulphurization, heavy metal leaching and bioconversion of crude oils. Thermostable enzymes such as DNA polymerase, amylases, xylanases proteases and lipase are required in basic research and biotechnology. Organic solvent tolerant bacteria that exhibit the ability to degrade crude oil, polyaromatic hydrocarbons, or cholesterol or can utilize sulphure compounds have been isolated. e.g. Strain Y-40 degrades hydrocarbons and belongs to Candida. It can grow well in the presence of solvents such as n-octane, isooctane, cyclooctane, or kerosene at a concentration of 50 % (v/v) the first strain that isolated for organic solvent tolerant bacterium e.g. Pseudomonas putida can grow in more than 50% toluene. Obligate extremophiles such as thiobacillus thiooxidans and thiobacillus ferrooxidanse occur in highly acidic environments. T. ferroxidans reduces sulphure compounds as well as ferrous ion to ferric. This leads to production of acids.
Currently these bacteria are utilized in the Biomining used in minerals leaching for example. T. ferroxidans used to isolate sulphure from the coal. Many extremophiles are used for craft industry for leather tanning (B. subtilis and B. licheniformis) with the help of various alkalophiles. Alkalophiles such as bacillus strains N-4 are used for cellulose digestion in the waste water treatment to digest cellulose. xylanases are produced by the alkalophiles Aeromonas strain 212 that can hydrolyze the beta 1.-4 linkage in the xylan polysaccharides in the crop plant. They show good activity and stability at pH 9. Neutral metalloproteases from B. amyloliquifaciens can be used in the brewing industry, when substituting malt with unmlated barley to increase the amount of free amino acids. The large-scale use of enzymes in solution Several enzymes, especially those used in starch processing, high-fructose syrup manufacture, textile desizing and detergent formulation, are now traded as commodity products on the world's markets. Although the cost of enzymes for use at the research scale is often very high, where there is a clear large-scale need for an enzyme its relative cost reduces dramatically with increased production. Relatively few enzymes, notably those in detergents, meat tenderizers and garden composting agents, are sold directly to the public. Most are used by industry to produce improved or novel products, to bypass long and involved chemical synthetic pathways or for use in the separation and purification of isomeric mixtures. Many of the most useful, but least-understood, uses of free enzymes are in the food industry. Here they are used, together with endogenous enzymes, to produce or process foodstuffs, which are only rarely substantially refined. Their action, however apparently straightforward, is complicated due to the effect that small amounts of by-products or associated reaction products have on such subjective effects as taste, smell, colour and texture. The use of enzymes in the non-food (chemicals and pharmaceuticals) sector is relatively straightforward. Products are generally separated and purified and, therefore, they are not prone to the subtleties available to food products. Most such enzymic conversions benefit from the use of immobilised enzymes or biphasic systems The use of enzymes in detergents The use of enzymes in detergent formulations is now common in developed countries, with over half of all detergents presently available containing enzymes. In spite of the fact that the detergent industry is the largest single market for enzymes at 25 - 30% of total sales. details of the enzymes used and the ways in which they are used, have rarely been published. Dirt comes in many forms and includes proteins, starches and lipids. In addition, clothes that have been starched must be freed of the starch. Using detergents in water at high temperatures and with vigorous mixing, it is possible to remove most types of dirt but the cost of heating the water is high and lengthy mixing or beating will shorten the life of clothing and other materials. The use of enzymes allows lower temperatures to be employed and shorter periods of agitation are needed, often after a preliminary period of soaking. In general, enzyme detergents remove protein from clothes soiled with blood, milk, sweat, grass, etc. far more effectively than non-enzyme detergents. However, using modern bleaching and brightening agents, the difference between looking clean and being clean may be difficult to discern. At present only proteases and amylases are commonly used. Although a wide range of lipases is known, it is only very recently that lipases suitable for use in detergent preparations have been described. Detergent enzymes must be cost-effective and safe to use. Early attempts to use proteases foundered because of producers and users developing hypersensitivity. This was combatted by developing dust-free granulates (about 0.5 mm in diameter) in which the enzyme is incorporated into an inner core, containing inorganic salts (e.g. NaCI) and sugars as preservative, bound with reinforcing, fibres of carboxymethyl cellulose or similar protective colloid. This core is coated with inert waxy materials made from paraffin oil or polyethylene glycol plus various
hydrophilic binders, which later disperse in the wash. This combination of materials both prevents dust formation and protects the enzymes against damage by other detergent components during storage. Enzymes are used in surprisingly small amounts in most detergent preparations, only 0.4 - 0.8% crude enzyme by weight (about 1% by cost). It follows that the ability to withstand the conditions of use is a more important criterion than extreme cheapness. Once released from its granulated form the enzyme must withstand anionic and non-ionic detergents, soaps, oxidants such as sodium perborate which generate hydrogen peroxide, optical brighteners and various lessreactive materials (Table 15.4), all at pH values between 8.0 and 10.5. Although one effect of incorporating enzymes is that lower washing temperatures may be employed with consequent savings in energy consumption, the enzymes must retain activity up to 60C. ________________________________________ Table 15.4 Compositions of an enzyme detergent Constituent Composition (%) Sodium tripolyphosphate (water softener, loosens dirt)a 38.0 Sodium alkane sulphonate (surfactant) 25.0 Sodium perborate tetrahydrate (oxidising agent)25.0 Soap (sodium alkane carboxylates) 3.0 Sodium sulphate (filler, water softener) 2.5 Sodium carboxymethyl cellulose (dirt-suspending agent) 1.6 Sodium metasilicate (binder, loosens dirt) 1.0 Bacillus protease (3% active) 0.8 Fluorescent brighteners 0.3 Foam-controlling agents Trace Perfume Trace Water to 100% a A recent trend is to reduce this phosphate content for environmental reasons. It may be replaced by sodium carbonate plus extra protease. ________________________________________ The enzymes used are all produced using species of Bacillus, mainly by just two companies. Novo Industri A/S produce and supply three proteases, Alcalase, from B. licheniformis, Esperase, from an alkalophilic strain of a B. licheniformis and Savinase, from an alkalophilic strain of B. amyloliquefaciens (often mistakenly attributed to B. subtilis). GistBrocades produce and supply Maxatase, from B. licheniformis. Alcalase and Maxatase (both mainly subtilisin) are recommended for use at 10-65C and pH 7-10.5. Savinase and Esperase may be used at up to pH 11 and 12, respectively. The -amylase supplied for detergent use is Termamyl, the enzyme from B. licheniformis which is also used in the production of glucose syrups. -Amylase is particularly useful in dish-washing and de-starching detergents. In addition to the granulated forms intended for use in detergent powders, liquid preparations in solution in water and slurries of the enzyme in a non-ionic surfactant are available for formulating in liquid 'spotting' concentrates, used for removing stubborn stains. Preparations containing both Termamyl and Alcalase are produced, Termamyl being sufficiently resistant to proteolysis to retain activity for long enough to fulfil its function. It should be noted that all the proteolytic enzymes described are fairly nonspecific serine endoproteases, giving preferred cleavage on the carboxyl side of hydrophobic amino acid residues but capable of hydrolysing most peptide links. They convert their substrates into small, readily soluble fragments which can be removed easily from fabrics. Only serine protease; may be used in detergent formulations: thiol proteases (e.g. papain) would be oxidised by the bleaching agents, and metalloproteases (e.g. thermolysin) would lose their metal cofactors due to complexing with the water softening agents or hydroxyl ions. The enzymes are supplied in forms (as described above) suitable for formulation by
detergent manufacturers. Domestic users are familiar with powdered preparations but liquid preparations for home use are increasingly available. Household laundering presents problems quite different from those of industrial laundering: the household wash consists of a great variety of fabrics soiled with a range of materials and the user requires convenience and effectiveness with less consideration of the cost. Home detergents will probably include both an amylase and a protease and a lengthy warm-water soaking time will be recommended. Industrial laundering requires effectiveness at minimum cost so heated water will be re-used if possible. Large laundries can separate their 'wash' into categories and thus minimise the usage of water and maximise the effectiveness of the detergents. Thus white cotton uniforms from an abattoir can be segregated for washing, only protease being required. A pre-wash soaking for 10-20 min at pH up to 11 and 30-40C is followed by a main wash for 10-20 min at pH 11 and 60-65C. The water from these stages is discarded to the sewer. A third wash includes hypochlorite as bleach which would inactivate the enzymes rapidly. The water from this stage is used again for the pre-wash but, by then, the hypochlorite concentration is insufficient to harm the enzyme. This is essentially a batch process: hospital laundries may employ continuous washing machines, which transfer less-initially-dirty linen from a pre-rinse initial stage, at 32C and pH 8.5, into the first wash at 60C and pH 11, then to a second wash, containing hydrogen peroxide, at 71C and pH 11, then to a bleaching stage and rinsing. Apart from the pre-soak stage, from which water is run to waste, the process operates countercurrently. Enzymes are used in the pre-wash and in the first wash, the levels of peroxide at this stage being insufficient to inactivate the enzymes. There are opportunities to extend the use of enzymes in detergents both geographically and numerically. They have not found widespread use in developing countries which are often hot and dusty, making frequent washing of clothes necessary. The recent availability of a suitable lipase may increase the quantities of enzymes employed very significantly. There are, perhaps, opportunities for enzymes such as glucose oxidase, lipoxygenase and glycerol oxidase as means of generating hydrogen peroxide in situ. Added peroxidases may aid the bleaching efficacy of this peroxide. A recent development in detergent enzymes has been the introduction of an alkaline-stable fungal cellulase preparation for use in washing cotton fabrics. During use, small fibres are raised from the surface of cotton thread, resulting in a change in the 'feel' of the fabric and, particularly, in the lowering of the brightness of colours. Treatment with cellulase removes the small fibres without apparently damaging the major fibres and restores the fabric to its 'as new' condition. The cellulase also aids the removal of soil particles from the wash by hydrolyzing associated cellulose fibres. 15.12 Applications of proteases in the food industry Certain proteases have been used in food processing for centuries and any record of the discovery of their activity has been lost in the mists of time. Rennet (mainly chymosin), obtained from the fourth stomach (abomasum) of unweaned calves has been used traditionally in the production of cheese. Similarly, papain from the leaves and unripe fruit of the pawpaw (Carica papaya) has been used to tenderize meats. These ancient discoveries have led to the development of various food applications for a wide range of available proteases from many sources, usually microbial. Proteases may be used at various pH values, and they may be highly specific in their choice of cleavable peptide links or quite non-specific. Proteolysis generally increases the solubility of proteins at their isoelectric points. 15.13 Rennet in cheese making The action of rennet in cheese making is an example of the hydrolysis of a specific peptide linkage, between phenylalanine and methionine residues (-Phe105Met106-) in the -casein protein present in milk . The -casein acts by stabilizing the colloidal nature of the milk, its hydrophobic N-terminal region associating with the lipophilic regions of the otherwise insoluble - and -
casein molecules, whilst its negatively charged C-terminal region associates with the water and prevents the casein micelles from growing too large. Hydrolysis of the labile peptide linkage between these two domains, resulting in the release of a hydrophilic glycosylated and phosphorylated oligopeptide (caseino macropeptide) and the hydrophobic para--casein, removes this protective effect, allowing coagulation of the milk to form curds, which are then compressed and turned into cheese (Figure 4.1). The coagulation process depends upon the presence of Ca2+ and is very temperature dependent (Q10 = 11) and so can be controlled easily. Calf rennet, consisting of mainly chymosin with a small but variable proportion of pepsin, is a relatively expensive enzyme and various attempts have been made to find cheaper alternatives from microbial sources These have ultimately proved to be successful and microbial rennets are used for about 70% of US cheese and 33% of cheese production world-wide. ________________________________________ Figure 15. 2 Outline method for the preparation of cheese. ________________________________________ The major problem that had to be overcome in the development of the microbial rennets was temperature lability. Chymosin is a relatively unstable enzyme and once it has done its major job, little activity remains. However, the enzyme from Mucor miehei retains activity during the maturation stages of cheese-making and produces bitter off-flavours. Treatment of the enzyme with oxidising agents (e.g. H2O2, peracids), which convert methionine residues to their sulfoxides, reduces its thermostability by about 10C and renders it more comparable with calf rennet. This is a rare example of enzyme technology being used to destabilise an enzyme Attempts have been made to clone chymosin into Escherichia coli and Saccharomyces cerevisiae but, so far, the enzyme has been secreted in an active form only from the latter. The development of unwanted bitterness in ripening cheese is an example of the role of proteases in flavour production in foodstuffs. The action of endogenous proteases in meat after slaughter is complex but 'hanging' meat allows flavour to develop, in addition to tenderising it. It has been found that peptides with terminal acidic amino acid residues give meaty, appetising flavours akin to that of monosodium glutamate. Non-terminal hydrophobic amino acid residues in medium-sized oligopeptides give bitter flavours, the bitterness being less intense with smaller peptides and disappearing altogether with larger peptides. Application of this knowledge allows the tailoring of the flavour of protein hydrolysates. The presence of proteases during the ripening of cheeses is not totally undesirable and a protease from Bacillus amyloliquefaciens may be used to promote flavour production in cheddar cheese. Lipases from Mucor miehei or Aspergillus niger are sometimes used to give stronger flavours in Italian cheeses by a modest lipolysis, increasing the amount of free butyric acid. They are added to the milk (30 U l-1) before the addition of the rennet. When proteases are used to depolymerise proteins, usually non-specifically, the extent of hydrolysis (degree of hydrolysis) is described in DH units where: (15.1) Commercially, using enzymes such as subtilisin, DH values of up to 30 are produced using protein preparations of 8-12% (w/w). The enzymes are formulated so that the value of the enzyme : substrate ratio used is 2-4% (w/w). At the high pH needed for effective use of subtilisin, protons are released during the proteolysis and must be neutralised: subtilisin (pH 8.5) H2N-aa-aa-aa-aa-aa-COO- H2N-aa-aa-aa-COO- + H2N-aa-aa-COO- + H+ [15.2] where aa is an amino acid residue. Correctly applied proteolysis of inexpensive materials such as soya protein can increase the range and value of their usage, as indeed occurs naturally in the production of soy sauce. Partial hydrolysis of soya protein, to around 3.5 DH greatly increases its 'whipping expansion', further hydrolysis, to around 6 DH
improves its emulsifying capacity. If their flavours are correct, soya protein hydrolysates may be added to cured meats. Hydrolysed proteins may develop properties that contribute to the elusive, but valuable, phenomenon of 'mouth feel' in soft drinks. Proteases are used to recover protein from parts of animals (and fish) would otherwise go to waste after butchering. About 5% of the meat can be removed mechanically from bone. To recover this, bones are mashed incubated at 60C with neutral or alkaline proteases for up to 4 h. The meat slurry produced is used in canned meats and soups. Large quantities of blood are available but, except in products such black puddings, they are not generally acceptable in foodstuffs because of their colour. The protein is of a high quality nutritionally and is de-haemed using subtilisin. Red cells are collected and haemolysed in water. Subtilisin is added and hydrolysis is allowed to proceed batchwise, with neutralization of the released protons, to around 18 DH, when the hydrophobic haem molecules precipitate. Excessive degradation is avoided to prevent the formation of bitter peptides. The enzyme is inactivated by a brief heat treatment at 85C and the product is centrifuged; no residual activity allowed into meat products. The haem-containing precipitate is recycled and the light-brown supernatant is processed through activated carbon beads to remove any residual haem. The purified hydrolysate, obtained in 60% yield, may be spray-dried and is used in cured meats, sausages and luncheon meats. 15.14 Meat tenderization Meat tenderisation is the softening of the meat for uses in the food by the endogenous proteases in the muscle after slaughter is a complex process which varies with the nutritional, physiological and even psychological (i.e. frightened or not) state of the animal at the time of slaughter. Meat of older animals remains tough but can be tenderised by injecting inactive papain into the jugular vein of the live animals shortly before slaughter. Injection of the active enzyme would rapidly kill the animal in an unacceptably painful manner so the inactive oxidised disulfide form of the enzyme is used. On slaughter, the resultant reducing conditions cause free thiols to accumulate in the muscle, activating the papain and so tenderising the meat. This is a very effective process as only 2 - 5 ppm of the inactive enzyme needs to be injected. Recently, however, it has found disfavour as it destroys the animals heart, liver and kidneys that otherwise could be sold and, being reasonably heat stable, its action is difficult to control and persists into the cooking process. 15.15 Baking industry Proteases are also used in the baking industry. Where appropriate, dough may be prepared more quickly if its gluten is partially hydrolysed. A heat-labile fungal protease is used so that it is inactivated early in the subsequent baking. Weakgluten flour is required for biscuits in order that the dough can be spread thinly and retain decorative impressions. In the past this has been obtained from European domestic wheat but this is being replaced by high-gluten varieties of wheat. The gluten in the flour derived from these must be extensively degraded if such flour is to be used efficiently for making biscuits or for preventing shrinkage of commercial pie pastry away from their aluminium dishes. 15.16 The use of enzymes in starch hydrolysis Starch is the commonest storage carbohydrate in plants. It is used by the plants themselves, by microbes and by higher organisms so there is a great diversity of enzymes able to catalyse its hydrolysis. Starch from all plant sources occurs in the form of granules which differ markedly in size and physical characteristics from species to species. Chemical differences are less marked. The major difference is the ratio of amylose to amylopectin; e.g. corn starch from waxy maize contains only 2% amylose but that from amylomaize is about 80% amylose. Some starches, for instance from potato, contain covalently bound phosphate in small amounts (0.2% approximately), which has significant effects on the physical properties of the starch but does not interfere with its hydrolysis. Acid
hydrolysis of starch has had widespread use in the past. It is now largely replaced by enzymic processes, as it required the use of corrosion resistant materials, gave rise to high colour and saltash content (after neutralisation), needed more energy for heating and was relatively difficult to control. ________________________________________ Figure 15. 3 The use of enzymes in processing starch. Typical conditions are given. ________________________________________ Of the two components of starch, amylopectine presents the great challenge to hydrolytic enzyme systems. This is due to the residues involved in -1,6glycosidic branch points which constitute about 4 - 6% of the glucose present. Most hydrolytic enzymes are specific for -1,4-glucosidic links yet the -1,6glucosidic links must also be cleaved for complete hydrolysis of amylopectin to glucose. Some of the most impressive recent exercises in the development of new enzymes have concerned debranching enzymes. It is necessary to hydrolyse starch in a wide variety of processes which may be condensed into two basic classes: Processes in which the starch hydrolysate is to be used by microbes or man, and Processes in which it is necessary to eliminate starch. In the former processes, such as glucose syrup production, starch is usually the major component of reaction mixtures, whereas in the latter processes, such as the processing of sugar cane juice, small amounts of starch which contaminate nonstarchy materials are removed. Enzymes of various types are used in these processes. Although starches from diverse plants may be utilised, corn is the world's most abundant source and provides most of the substrate used in the preparation of starch hydrolysates. There are three stages in the conversion of starch (Figure 15.3): gelatinization, involving the dissolution of the nano-gram-sized starch granules to form a viscous suspension; liquefaction, involving the partial hydrolysis of the starch, with concomitant loss in viscosity; and Saccharification, involving the production of glucose and maltose by further hydrolysis. a. Gelatinization is achieved by heating starch with water, and occurs necessarily and naturally when starchy foods are cooked. Gelatinized starch is readily liquefied by partial hydrolysis with enzymes or acids and saccharified by further acidic or enzymic hydrolysis. The starch and glucose syrup industry uses the expression dextrose equivalent or DE, similar in definition to the DH units of proteolysis, to describe its products, where: (15 .3) In practice, this is usually determined analytically by use of the closely related, but not identical, expression: (15.4) Thus, DE represents the percentage hydrolysis of the glycosidic linkages present. Pure glucose has a DE of 100, pure maltose has a DE of about 50 (depending upon the analytical methods used; see equation (15.4)) and starch has a DE of effectively zero. During starch hydrolysis, DE indicates the extent to which the starch has been cleaved. Acid hydrolysis of starch has long been used to produce 'glucose syrups' and even crystalline glucose (dextrose monohydrate). Very considerable amounts of 42 DE syrups are produced using acid and are used in many applications in confectionery. Further hydrolysis using acid is not satisfactory because of undesirably coloured and flavoured breakdown products. Acid hydrolysis appears to be a totally random process which is not influenced by the presence of -1,6-glucosidic linkages. ________________________________________ Table 15.5 Enzymes used in starch hydrolysis
Enzyme EC number Source Action -Amylase 3.2.1.1 Bacillus amyloliquefaciens Only -1,4-oligosaccharide links are cleaved to give -dextrins and predominantly maltose (G2), G3, G6 and G7 oligosaccharides B. licheniformis Only 1,4-oligosaccharide links are cleaved to give -dextrins and predominantly maltose, G3, G4 and G5 oligosaccharides Aspergillus oryzae, A. niger Only -1,4 oligosaccharide links are cleaved to give -dextrins and predominantly maltose and G3 oligosaccharides Saccharifying -amylase 3.2.1.1 B. subtilis (amylosacchariticus) Only -1,4-oligosaccharide links are cleaved to give -dextrins with maltose, G3, G4 and up to 50% (w/w) glucose -Amylase 3.2.1.2 Malted barley Only -1,4-links are cleaved, from nonreducing ends, to give limit dextrins and -maltose Glucoamylase 3.2.1.3 A. niger -1,4 and 1,6-links are cleaved, from the nonreducing ends, to give -glucose Pullulanase 3.2.1.41 B. acidopullulyticus Only -1,6-links are cleaved to give straight-chain maltodextrins ________________________________________ The nomenclature of the enzymes used commercially for starch hydrolysis The nomenclature of the enzymes used commercially for starch hydrolysis is somewhat confusing and the EC numbers sometimes lump together enzymes with subtly different activities (Table 15.5). For example, -amylase may be subclassified as liquefying or saccharifying amylases but even this classification is inadequate to encompass all the enzymes that are used in commercial starch hydrolysis. One reason for the confusion in the nomenclature is the use of the anomeric form of the released reducing group in the product rather than that of the bond being hydrolysed; the products of bacterial and fungal -amylases are in the configuration and the products of -amylases are in the -configuration, although all these enzymes cleave between -1,4-linked glucose residues. The -amylases (1,4--D-glucan glucanohydrolases) are endohydrolases which cleave 1,4--D-glycosidic bonds and can bypass but cannot hydrolyze 1,6--D-glucosidic branch points. Commercial enzymes used for the industrial hydrolysis of starch are produced by Bacillus amyloliquefaciens (supplied by various manufacturers) and by B. licheniformis (supplied by Novo Industri A/S as Termamyl). They differ principally in their tolerance of high temperatures, Termamyl retaining more activity at up to 110C, in the presence of starch, than the B. amyloliquefaciens -amylase. The maximum DE obtainable using bacterial -amylases is around 40 but prolonged treatment leads to the formation of maltulose (4--D-glucopyranosyl-Dfructose), which is resistant to hydrolysis by glucoamylase and -amylases. DE values of 8-12 are used in most commercial processes where further saccharification is to occur. The principal requirement for liquefaction to this extent is to reduce the viscosity of the gelatinised starch to ease subsequent processing. Different approaches for starch liquefaction Various manufacturers use different approaches to starch liquefaction using amylases but the principles are the same. Granular starch is slurried at 30-40% (w/w) with cold water, at pH 6.0-6.5, containing 20-80 ppm Ca2+ (which stabilizes and activates the enzyme) and the enzyme is added (via a metering pump). The amylase is usually supplied at high activities so that the enzyme dose is 0.5-0.6 kg tonne-1 (about 1500 U kg-1 dry matter) of starch. When Termamyl is used, the slurry of starch plus enzyme is pumped continuously through a jet cooker, which is heated to 105C using live steam. Gelatinization occurs very rapidly and the enzymic activity, combined with the significant shear forces, begins the hydrolysis. The residence time in the jet cooker is very brief. The partly gelatinized starch is passed into a series of holding tubes maintained at 100105C and held for 5 min to complete the gelatinization process. Hydrolysis to the required DE is completed in holding tanks at 90-100C for 1 to 2 h. These tanks contain baffles to discourage backmixing. Similar processes may be used with B.
amyloliquefaciens -amylase but the maximum temperature of 95C must not be exceeded. This has the drawback that a final 'cooking' stage must be introduced when the required DE has been attained in order to gelatinize the recalcitrant starch grains present in some types of starch which would otherwise cause cloudiness in solutions of the final product. Saccharification The liquefied starch is usually saccharified but comparatively small amounts are spray-dried for sale as 'maltodextrins' to the food industry mainly for use as bulking agents and in baby food. In this case, residual enzymic activity may be destroyed by lowering the pH towards the end of the heating period. Fungal -amylase also finds use in the baking industry. It often needs to be added to bread-making flours to promote adequate gas production and starch modification during fermentation. This has become necessary since the introduction of combine harvesters. They reduce the time between cutting and threshing of the wheat, which previously was sufficient to allow a limited sprouting so increasing the amounts of endogenous enzymes. The fungal enzymes are used rather than those from bacteria as their action is easier to control due to their relative heat lability, denaturing rapidly during baking. The liquefied starch at 8 -12 DE is suitable for saccharification to produce syrups with DE values of from 45 to 98 or more. The greatest quantities produced are the syrups with DE values of about 97. At present these are produced using the exoamylase, glucan 1,4--glucosidase (1,4--D-glucan glucohydrolase, commonly called glucoamylase but also called amyloglucosidase and -amylase), which releases -D-glucose from 1,4--, 1,6-- and 1,3--linked glucans. In theory, carefully liquefied starch at 8 -12 DE can be hydrolysed completely to produce a final glucoamylase reaction mixture with DE of 100 but, in practice, this can be achieved only at comparatively low substrate concentrations. The cost of concentrating the product by evaporation decreases that a substrate concentration of 30% is used. It follows that the maximum DE attainable is 96 - 98 with syrup composition 95 - 97% glucose, 1 - 2% maltose and 0.5 - 2% (w/w) isomaltose (-D-glucopyranosyl-(1,6)-D-glucose). This material is used after concentration, directly for the production of high-fructose syrups or for the production of crystalline glucose. Whereas liquefaction is usually a continuous process, saccharification is most often conducted as a batch process. The glucoamylase most often used is produced by Aspergillus niger strains. This has a pH optimum of 4.0 - 4.5 and operates most effectively at 60C, so liquefied starch must be cooled and its pH adjusted before addition of the glucoamylase. The cooling must b rapid, to avoid retrogradation (the formation of intractable insoluble aggregates of amylose; the process that gives rise to the skin on custard). Any remaining bacterial -amylase will be inactivated when the pH is lowered; however, this may be replaced later by some acid-stable -amylase which is normally present in the glucoamylase preparations. When conditions are correct the glucoamylase is added, usually at the dosage of 0.65 - 0.80 litre enzyme preparation.tonne-1 starch (200 U kg-1). Saccharification is normally conducted in vast stirred tanks, which may take several hours to fill (and empty), so time will be wasted if the enzyme is added only when the reactors are full. The alternatives are to meter the enzyme at a fixed ratio or to add the whole dose of enzyme at the commencement of the filling stage. The latter should give the most economical use of the enzyme. ________________________________________ Figure 15. 4. The % glucose formed from 30% (w/w) 12 DE maltodextrin, at 60C and pH 4.3, using various enzyme solutions. 200 U kg-1 Aspergillus niger glucoamylase; -----------400 U kg-1 A. niger glucoamylase; 200 U kg-1 A. niger glucoamylase plus 200 U kg-1 Bacillus acidopullulyticus pullulanase. The relative improvement on the addition of pullulanase is even greater at higher substrate concentrations. ________________________________________ The saccharification process takes about 72 h to complete but may, of course, be
speeded up by the use of more enzyme. Continuous saccharification is possible and practicable if at least six tanks are used in series. It is necessary to stop the reaction, by heating to 85C for 5 min, when a maximum DE has been attained. Further incubation will result in a fall in the DE, to about 90 DE eventually, caused by the formation of isomaltose as accumulated glucose re-polymerises with the approach of thermodynamic equilibrium (Figure 15.4). Final step in syrup formation The saccharified syrup is filtered to remove fat and denatured protein released from the starch granules and may then be purified by passage through activated charcoal and ion-exchange resins. It should be remembered that the dry substance concentration increases by about 11 % during saccharification, because one molecule of water is taken up for each glycosidic bond hydrolysed (molecule of glucose produced). Although glucoamylase catalyses the hydrolysis of 1,6--linkages, their breakdown is slow compared with that of 1,4--linkages (e.g. the rates of hydrolyzing the 1,4-, 1,6- and 1,3--links in tetrasaccharides are in the proportions 300 : 6 : 1). It is clear that the use of a debranching enzyme would speed the overall saccharification process but, for industrial use such an enzyme must be compatible with glucoamylase. Two types of debranching enzymes are available: pullulanase, which acts as an exo hydrolase on starch dextrins; and isoamylase (EC.3.2.1.68), which is a true endohydrolase. Novo Industri A/S have recently introduced a suitable pullulanase, produced by a strain of Bacillus acidopullulyticus. The pullulanase from Klebsiella aerogenes which has been available commercially to some time is unstable at temperatures over 45C but the B. acidopullulyticus enzymes can be used under the same conditions as the Aspergillus glucoamylase (60C, pH 4.0-4.5). The practical advantage of using pullulanase together with glucoamylase is that less glucoamylase need be used This does not in itself give any cost advantage but because less glucoamylase is used and fewer branched oligosaccharides accumulate toward the end of the saccharification, the point at which isomaltose production becomes significant occurs at higher DE (Figure 4.3). It follows that higher DE values and glucose contents can be achieved when pullulanase is use (98 - 99 DE and 95 - 97% (w/w) glucose, rather than 97 - 98 DE) and higher substrate concentrations (30 - 40% dry solids rather than 25 - 30%) may be treated. The extra cost of using pullulanase is recouped by savings in evaporation and glucoamylase costs. In addition, when the product is to be used to manufacture high-fructose syrups, there is a saving in the cost of further processing. The development of the B. acidopullulyticus pullulanase is an excellent example of what can be done if sufficient commercial pull exists for a new enzyme. The development of a suitable -D-glucosidase, in order to reduce the reversion, would be an equally useful step for industrial glucose production. Screening of new strains of bacteria for a novel enzyme of this type is a major undertaking. It is not surprising that more details of the screening procedures used are not readily available. 15.17 Production of syrups containing maltose Traditionally, syrups containing maltose as a major component have been produced by treating barley starch with barley -amylase. -Amylases (1,4--D-glucan maltohydrolases) are exohydrolases which release maltose from 1,4--linked glucans but neither bypass nor hydrolyse 1,6--linkages. High-maltose syrups (40 - 50 DE, 45-60% (w/w) maltose, 2 - 7% (w/w) glucose) tend not to crystallise, even below 0C and are relatively non-hygroscopic. They are used for the production of hard candy and frozen deserts. High conversion syrups (60 - 70 DE, 30 - 37% maltose, 35 - 43% glucose, 10% maltotriose, 15% other oligosaccharides, all by weight) resist crystallisation above 4C and are sweeter (Table 4.3). They are used for soft candy and in the baking, brewing and soft drinks industries. It might be expected that -amylase would be used to produce maltose-rich syrups from corn starch, especially as the combined action of -amylase and pullulanase give almost quantitative yields of maltose. This is not done on a significant scale nowadays
because presently available -amylases are relatively expensive, not sufficiently temperature stable (although some thermostable -amylases from species of Clostridium have recently been reported) and are easily inhibited by copper and other heavy metal ions. Instead fungal -amylases, characterised by their ability to hydrolyse maltotriose (G3) rather than maltose (G2) are employed often in combination with glucoamylase. Presently available enzymes, however, are not totally compatible; fungal -amylases requiring a pH of not less than 5.0 and a reaction temperature not exceeding 55C. High-maltose syrups (see Figure 15.3) are produced from liquefied starch of around 11 DE at a concentration of 35% dry solids using fungal -amylase alone. Saccharification occurs over 48 h, by which time the fungal -amylase has lost its activity. Now that a good pullulanase is available, it is possible to use this in combination with fungal -High-conversion syrups are produced using combinations of fungal -amylase and glucoamylase. These may be tailored to customers' specifications by adjusting the activities of the two enzymes used but inevitably, as glucoamylase is employed, the glucose content of the final product will be higher than that of high-maltose syrups. The stability of glucoamylase necessitates stopping the reaction, by heating, when the required composition is reached. It is now possible to produce starch hydrolysates with any DE between 1 and 100 and with virtually any composition using combinations of bacterial amylases, fungal -amylases, glucoamylase and pullulanase. ________________________________________ Table 15.6 The relative sweetness of food ingredients Food ingredient Relative sweetness (by weight, solids) Sucrose 1.0 Glucose 0.7 Fructose 1.3 Galactose 0.7 Maltose 0.3 Lactose 0.2 Raffinose 0.2 Hydrolysed sucrose 1.1 Hydrolysed lactose 0.7 Glucose syrup 11 DE <0.1 Glucose syrup 42 DE 0.3 Glucose syrup 97 DE 0.7 Maltose syrup 44 DE 0.3 High-conversion syrup 65 DE 0.5 HFCS (42% fructose)a 1.0 HFCS (55% fructose) 1.1 Aspartame 180 a HFCS, high-fructose corn syrup. 15.18 Enzymes in the sucrose industry The sucrose industry is a comparatively minor user of enzymes but provides few historically significant and instructive examples of enzyme technology The hydrolysis ('inversion') of sucrose, completely or partially, to glucose and fructose provides sweet syrups that are more stable (i.e. less likely crystallise) than pure sucrose syrups. The most familiar 'Golden Syrup' produced by acid hydrolysis of one of the less pure streams from the cane sugar refinery but other types of syrup are produced using yeast (Saccharomyces cerevisiae) invertase. Although this enzyme is unusual in that it suffers from substrate inhibition at high sucrose levels ( > 20% (w/w)), this does not prevent its commercial use at even higher concentrations: Figure 15. 5Traditionally, Invertase was produced on site by autolysing yeast cells. The autolysate was added to the syrup (70% sucrose (w/w)) to be inverted
together with small amounts of xylene to prevent microbial growth. Inversion was complete in 48 - 72 h at 50C and pH 4.5. The enzyme and xylene were removed during the subsequent refining and evaporation. Partially inverted syrups were (and still are) produced by blending totally inverted syrups with sucrose syrups. Now, commercially produced invertase concentrates are employed. The production of hydrolysates of a low molecular weight compound in essentially pure solution seems an obvious opportunity for the use of an immobilised enzyme, yet this is not done on a significant scale, probably because of the extreme simplicity of using the enzyme in solution and the basic conservatism of the sugar industry. Invertase finds another use in the production of confectionery with liquid or soft centres. These centers are formulated using crystalline sucrose and tiny (about 100 U kg-1, 0.3 ppm (w/w)) amounts of invertase. At this level of enzyme, inversion of sucrose is very slow so the centre remains solid long enough for enrobing with chocolate to be completed. Then, over a period of days or weeks, sucrose hydrolysis occurs and the increase in solubility causes the centers to become soft or liquid, depending on the water content of the centre preparation. Other enzymes are used as aids to sugar production and refining by removing materials which inhibit crystallisation or cause high viscosity. In some parts of the world, sugar cane contains significant amounts of starch, which becomes viscous, thus slowing filtration processes and making the solution hazy when the sucrose is dissolved. This problem can be overcome by using the most thermostable -amylases (e.g. Termamyl at about 5 U kg-1) which are entirely compatible with the high temperatures and pH values that prevail during the initial vacuum evaporation stage of sugar production. Other problems involving dextran and raffinose required the development of new industrial enzymes. A Dextran is produced by the action of dextransucrase (EC 2.4.1.5) from Leuconostoc mesenteroides on sucrose and found as a slime on damaged cane and beet tissue, especially when processing has been delayed in hot and humid climates. Raffinose, which consists of sucrose with -galactose attached through its C-1 atom to the 6 position on the glucose residue, is produced at low temperatures in sugar beet. Both dextran and raffinose have the sucrose molecule as part of their structure and both inhibit sucrose crystal growth. This produces plate-like or needle-like crystals which are not readily harvested by equipment designed for the approximately cubic crystals otherwise obtained. Dextran can produce extreme viscosity it process streams and even bring plant to a stop. Extreme dextran problems arc frequently solved by the use of fungal dextranases produced from Penicillium species. These are used (e.g. 10 U kg-1 raw juice, 55C, pH 5.5, 1 h) only in times of crisis as they are not sufficiently resistant to thermal denaturation for long-term use and are inactive at high sucrose concentrations. Because only small quantities are produced for use, this enzyme is relatively expensive. An enzyme sufficiently stable for prophylactic use would be required in order to benefit from economies of scale. Raffinose may be hydrolysed to galactose and sucrose by a fungal raffinase. 15.19 Glucose from cellulose There is very much more cellulose available, as a potential source of glucose, than starch, yet cellulose is not a significant source of pure glucose. The reasons for this are many, some technical, some commercial. The fundamental reason is that starch is produced in relatively pure forms by plants for use as an easily biodegradable energy and carbon store. Cellulose is structural and is purposefully combined and associated with lignin and pentosans, so as to resist biodegradation; dead trees take several years to decay even in tropical rainforests. A typical waste cellulolytic material contains less than half cellulose, most of the remainder consisting of roughly equal quantities of lignin and pentosans. A combination of enzymes is needed to degrade this mixture. These enzymes are comparatively unstable of low activity against native lignocellulose and subject
to both substrate and product inhibition. Consequently, although many cellulolytic enzymes exist and it is possible to convert pure cellulose to glucose using only enzymes, the cost of this conversion is excessive. The enzymes might be improved by strain selection from the wild or by mutation but problems caused by the physical nature of cellulose are not so amenable to solution. Granular starch is readily stirred in slurries containing 40% (w/v) solids and is easily solubilised but, even when pure, fibrous cellulose forms immovable cakes at 10% solids and remains insoluble in all but the most exotic (and enzyme denaturing) solvents. Impure cellulose often contains almost an equal mass of lignin, which is of little or no value as a by-product and is difficult an expensive to remove. Commercial cellulase preparations from Trichoderma reesei consist of mixtures of the synergistic enzymes: cellulase (EC 3.2.1.4), an endo-1,4-D -glucanase; glucan 1,4--glucosidase (EC 3.2.1.74), and exo-1,4--glucosidase; and cellulose 1,4--cellobiosidase (EC 3.2.1.91), an exo-cellobiohydrolase (see Figure 15.6). They are used for the removal of relatively small concentrations of cellulose complexes which have been found to interfere in the processing of plant material in, for example, the brewing and fruit juice industries. ________________________________________ Figure 15. 6 Outline of the relationship between the enzyme activities in the hydrolysis of cellulose. || represents inhibitory effects. Endo-1,4--glucanase is the rate-controlling activity and may consist of a mixture of enzymes acting on cellulose of different degrees of crystallinity. It acts synergistically with both exo-1,4--glucosidase and exo-cellobiohydrolase. Exo-1,4--glucosidase is a product-inhibited enzyme. Exo-cellobiohydrolase is product inhibited and additionally appears to be inactivated on binding to the surface of crystalline cellulose. ________________________________________ Proper economic analysis reveals that cheap sources of cellulose prove to be generally more expensive as sources of glucose than apparently more expensive starch. Relatively pure cellulose is valuable in its own right, as a paper pulp and chipboard raw material, which currently commands a price of over twice that of corn starch. With the increasing world shortage of pulp it cannot be seen realistically as an alternative source of glucose in the foreseeable future. Knowledge of enzyme systems capable of degrading lignocellulose is advancing rapidly but it is unlikely that lignocellulose will replace starch as a source of glucose syrups for food use. It is, however, quite possible that it may be used, in a process involving the simultaneous use of both enzymes and fermentative yeasts, to produce ethanol; the utilisation of the glucose by the yeast removing its inhibitory effect on the enzymes. It should be noted that cellobiose is a nonfermentable sugar and must be hydrolysed by additional -glucosidase (EC 3.2.1.21, also called cellobiase for maximum process efficiency (Figure 15.6). 15.20 The use of lactases in the dairy industry Lactose is present at concentrations of about 4.7% (w/v) in milk and the whey (supernatant) left after the coagulation stage of cheese-making. Its presence in milk makes it unsuitable for the majority of the world's adult population, particularly in those areas which have traditionally not had a dairy industry. Real lactose tolerance is confined mainly to peoples whose origins lie in Northern Europe or the Indian subcontinent and is due to 'lactase persistence'; the young of all mammals clearly are able to digest milk but in most cases this ability reduces after weaning. Of the Thai, Chinese and Black American populations, 97%, 90% and 73% respectively, are reported to be lactose intolerant, whereas 84% and 96% of the US White and Swedish populations, respectively, are tolerant. Additionally, and only very rarely some individuals suffer from inborn metabolic lactose intolerance or lactase deficiency, both of which may be noticed at birth.
The need for low-lactose milk is particularly important in food-aid programmes as severe tissue dehydration, diarrhoea and even death may result from feeding lactose containing milk to lactose-intolerant children and adults suffering from protein-calorie malnutrition. In all these cases, hydrolysis of the lactose to glucose and galactose would prevent the (severe) digestive problems. Another problem presented by lactose is its low solubility resulting in crystal formation at concentrations above 11 % (w/v) (4C). This prevents the use of concentrated whey syrups in many food processes as they have a unpleasant sandy texture and are readily prone to microbiological spoilage. Adding to this problem, the disposal of such waste whey is expensive (often punitively so) due to its high biological oxygen demand. These problems may be overcome by hydrolysis of the lactose in whey; the product being about four times as sweet (see Table 15.6), much more soluble and capable of forming concentrated, microbiologically secure, syrups (70% (w/v)). Figure 15. 7 Lactose may be hydrolysed by lactase, a -galactosidase. Commercially, it may be prepared from the dairy yeast Kluyveromyces fragilis (K. marxianus var. marxianus), with a pH optimum (pH 6.5-7.0) suitable for the treatment of milk, or from the fungi Aspergillus oryzae or A. niger, with pH optima (pH 4.5-6.0 and 3.0-4.0, respectively) more suited to whey hydrolysis. These enzymes are subject to varying degrees of product inhibition by galactose. In addition, at high lactose and galactose concentrations, lactase shows significant transferase ability and produces -1,6-linked galactosyl oligosaccharides. Lactases are now used in the production of ice cream and sweetened flavoured and condensed milks. When added to milk or liquid whey (2000 U kg-1) and left for about a day at 5C about 50% of the lactose is hydrolysed, giving a sweeter product which will not crystallise if condensed or frozen. This method enables otherwise-wasted whey to replace some or all of the skim milk powder used in traditional ice cream recipes. It also improves the 'scoopability' and creaminess of the product. Smaller amounts of lactase may be added to long-life sterilised milk to produce a relatively inexpensive lactose-reduced product (e.g. 20 U kg-1, 20C, 1 month of storage). Generally, however, lactase usage has not reached its full potential, as present enzymes are relatively expensive and can only be used at low temperatures. 15.21 Enzymes in the fruit juice, wine, brewing and distilling industries One of the major problems in the preparation of fruit juices and wine is cloudiness due primarily to the presence of pectins. These consist primarily of 1,4-anhydrogalacturonic acid polymers, with varying degrees of methyl esterification. They are associated with other plant polymers and, after homogenisation, with the cell debris. The cloudiness that they cause is difficult to remove except by enzymic hydrolysis. Such treatment also has the additional benefits of reducing the solution viscosity, increasing the volume of juice produced (e.g. the yield of juice from white grapes can be raised by 15%), subtle but generally beneficial changes in the flavour and, in the case of wine-making, shorter fermentation times. Insoluble plant material is easily removed by filtration, or settling and decantation, once the stabilising effect of the pectins on the colloidal haze has been removed. Commercial pectolytic enzyme preparations are produced from Aspergillus niger and consist of a synergistic mixture of enzymes: polygalacturonase (EC 3.2.1.15), responsible for the random hydrolysis of 1,4- D-galactosiduronic linkages; pectinesterase (EC 3.2.1.11), which releases methanol from the pectyl methyl esters, a necessary stage before the polygalacturonase can act fully (the increase in the methanol content of such treated juice is generally less than the natural
concentrations and poses no health risk); pectin lyase (EC 4.2.2.10), which cleaves the pectin, by an elimination reaction releasing oligosaccharides with non-reducing terminal 4-deoxymethyl--D-galact-4enuronosyl residues, without the necessity of pectin methyl esterase action; and hemicellulase (a mixture of hydrolytic enzymes including: xylan endo-1,3-xylosidase, EC 3.2.1.32; xylan 1,4--xylosidase, EC 3.2.1.37; and -Larabinofuranosidase, EC 3.2.1.55), strictly not a pectinase but its adventitious presence is encouraged in order to reduce hemicellulose levels. The optimal activity of these enzymes is at a pH between 4 and 5 and generally below 50C. They are suitable for direct addition to the fruit pulps at levels around 20 U l-1 (net activity). Enzymes with improved characteristics of greater heat stability and lower pH optimum are currently being sought. In brewing, barley malt supplies the major proportion of the enzyme needed for saccharification prior to fermentation. Often other starch containing material (adjuncts) are used to increase the fermentable sugar and reduce the relative costs of the fermentation. Although malt enzyme may also be used to hydrolyse these adjuncts, for maximum economic return extra enzymes are added to achieve their rapid saccharification. It not necessary nor desirable to saccharify the starch totally, as non-fermentable dextrins are needed to give the drink 'body' and stabilise its foam 'head'. For this reason the saccharification process is stopped, by boiling the 'wort', after about 75% of the starch has been converted into fermentable sugar. The enzymes used in brewing are needed for saccharification of starch (bacterial and fungal -amylases), breakdown of barley -1,4- and -1,3- linked glucan (glucanase) and hydrolysis of protein (neutral protease) to increase the (later) fermentation rate, particularly in the production of high-gravity beer, where extra protein is added. Cellulases are also occasionally used, particularly where wheat is used as adjunct but also to help breakdown the barley -glucans. Due to the extreme heat stability of the B. amyloliquefaciens -amylase, where this is used the wort must be boiled for a much longer period (e.g. 30 min) to inactivate it prior to fermentation. Papain is used in the later post-fermentation stages of beer-making to prevent the occurrence of protein- and tannin-containing 'chillhaze' otherwise formed on cooling the beer. Recently, 'light' beers, of lower calorific content, have become more popular. These require a higher degree of saccharification at lower starch concentrations to reduce the alcohol and total solids contents of the beer. This may be achieved by the use of glucoamylase and/or fungal -amylase during the fermentation. A great variety of carbohydrate sources are used world wide to produce distilled alcoholic drinks. Many of these contain sufficient quantities of fermentable sugar (e.g. rum from molasses and brandy from grapes), others contain mainly starch and must be saccharified before use (e.g. whiskey from barley malt, corn or rye). In the distilling industry, saccharification continues throughout the fermentation period. In some cases (e.g. Scotch malt whisky manufacture uses barley malt exclusively) the enzymes are naturally present but in others (e.g. grain spirits production) the more heat-stable bacterial -amylases may be used in the saccharification. 15.22 Glucose oxidase and catalase in the food industry Glucose oxidase is a highly specific enzyme, from the fungi Aspergillus niger and Penicillium, which catalyses the oxidation of -glucose to glucono-1,5-lactone (which spontaneously hydrolyses non-enzymically to gluconic acid) using molecular oxygen and releasing hydrogen peroxide. It finds uses in the removal of either glucose or oxygen from foodstuffs in order to improve their storage capability. Hydrogen peroxide is an effective bacteriocide and may be removed, after use, by treatment with catalase (derived from the same fungal fermentations as the glucose oxidase) which converts it to water and molecular oxygen: catalase 2H2O2 2H2O + O2 [4.4]
For most large-scale applications the two enzymic activities are not separated. Glucose oxidase and catalase may be used together when net hydrogen peroxide production is to be avoided. A major application of the glucose oxidase/catalase system is in the removal of glucose from egg-white before drying for use in the baking industry. A mixture of the enzymes is used (165 U kg-1) together with additional hydrogen peroxide (about 0.1 % (w/w)) to ensure that sufficient molecular oxygen is available, by catalase action, to oxidise the glucose. Other uses are in the removal of oxygen from the head-space above bottled and canned drinks and reducing non-enzymic browning in wines and mayonnaises.
Chapter-16
Enzyme Reactors
Introduction An enzyme reactor consists of a vessel, or series of vessels, used to perform a desired conversion by enzymic means. There are several important factors that determine the choice of reactor for a particular process. In general, the choice depends on the cost of a predetermined productivity within the product's specifications. This must be inclusive of the costs associated with substrate(s), downstream processing, labour, depreciation, overheads and process development, in addition to the more obvious costs concerned with building and running the enzyme reactor. Other contributing factors are the form of the enzyme of choice (i.e. free or immobilised), the kinetics of the reaction and the chemical and physical properties of an immobilization support including whether it is particulate, membranous or fibrous, and its density, compressibility, robustness, particle size and regenerability. Attention must also be paid to the scale of operation, the possible need for pH and temperature control, the supply and removal of gases and the stability of the enzyme, substrate and product. These factors will be
discussed in more detail with respect to the different types of reactor. There are two type of Reactor 1. Batch Reactor; 2. Continuous reactor Stirred tank batch reactor (STR). Batch membrane reactor (MR). Packed bed reactor (PBR). continuous flow stirred tank reactor (CSTR. continuous flow membrane reactor (CMR) fluidised bed reactor (FBR) 16.1 Batch Reactor Batch reactors generally consist of a tank containing a stirrer (stirred tank reactor, STR). Note that a batch reactor is one in which all of the product is removed, as rapidly as is practically possible, after a fixed time. The tank is normally fitted with fixed baffles that improve the stirring efficiency. Generally this means that the enzyme and substrate molecules must have identical residence times within the reactor, although in some circumstances there may be a need for further additions of enzyme and/or substrate (i.e. fed -batch operation). a. Disadvantage of using batch reactor 1. The operating costs of batch reactors are higher than for continuous processes due to the necessity for the reactors to be emptied and refilled both regularly and often. This procedure is not only expensive in itself but means that there are considerable periods when such reactors are not productive; it also makes uneven demands on both labour and services. 2. STRs can be used for processes involving non-immobilised enzymes, if the consequences of these contaminating the product are not severe. 3. Batch reactors also suffer from pronounced batch-to-batch variations, as the reaction conditions change with time, and may be difficult to scale-up, due to the changing power requirements for efficient fixing. b. Advantage of using Batch Bioreactor They do, however, have a number of advantageous features. Primary amongst these is their simplicity both in use and in process development. For this reason they are preferred for small-scale production of highly priced products, especially where the same equipment is to be used for a number of different conversions. They offer a closely controllable environment that is useful for slow reactions, where the composition may be accurately monitored, and conditions (e.g. temperature, pH, coenzyme concentrations) varied throughout the reaction. They are also of use when continuous operation of a process proves to be difficult due to the viscous or intractable nature of the reaction mix. ________________________________________ Figure 16. 1 Diagrams of various important enzyme reactor types. Stirred tank batch reactor (STR), which contains all of the enzyme and substrates) until the conversion is complete; batch membrane reactor (MR), where the enzyme is held within membrane tubes which allow the substrate to diffuse in and the product to diffuse out. This reactor may often be used in a semicontinuous manner, using the same enzyme solution for several batches; packed bed reactor (PBR), also called plug -flow reactor (PFR), containing a settled bed of immobilised enzyme particles; continuous flow stirred tank reactor (CSTR) which is a continuously operated version of (a); continuous flow membrane reactor (CMR) which is a continuously operated version of (b); fluidised bed reactor (FBR), where the flow of gas and/or substrate keeps the immobilised enzyme particles in a fluidised state. All reactors would additionally have heating/cooling coils (interior in reactors (a), and (d), and exterior, generally, in reactors (b), (c), (e) and (f)) and the stirred reactors may contain baffles in order to increase (reactors (a), (b), (d)
and (e) or decrease (reactor (f)) the stirring efficiency. The continuous reactors ((c) -(f)) may all be used in a recycle mode where some, or most, of the product stream is mixed with the incoming substrate stream. All reactors may use immobilised enzymes. In addition, reactors (a), (b) and (e) (plus reactors (d) and (f), if semipermeable membranes are used on their outlets) may be used with the soluble enzyme. ________________________________________ c. The expected productivity of a batch reactor The expected productivity of a batch reactor may be calculated by, assuming the validity of the non -reversible Michaelis -Menten reaction scheme with no diffusional control, inhibition or denaturation. The rate of reaction (v) may be expressed in terms of the volume of substrate solution within the reactor (VolS) and the time (t): (16.1) Therefore: (16.2) On integrating using the boundary condition that [S] = [S]0 at time (t) = 0: (16.3) Let the fractional conversion be X, where: (16.4) Therefore; (16.4a) and (16.4b) Also (16.4c) Therefore substituting using (5.4c) and (5.4b) in (5.3): (16.5) The change in fractional conversion and concentrations of substrate and product with time in a batch reactor is shown in Figure 16.2(a).
Figure 16. 2 This figure shows two related behaviours. (a) The change in substrate and product concentrations with time, in a batch reactor. The reaction S P is assumed, with the initial condition [S]0/Km = 10. The concentrations of substrate ( and product (-----------) are both normalised with respect to [S]0. The normalised time (i.e. t = t Vmax/[S]0) is relative to the time (t = 1) that would be required to convert all the substrate if the enzyme acted at Vmax throughout, the actual time for complete conversion being longer due to the reduction in the substrate concentration at the reaction progresses. The dashed line also indicates the variation of the fractional conversion (X) with t. (b) The change in substrate and product concentrations with reactor length for a PBR. The reaction S P is assumed with the initial condition, [S]0/Km = 10. The concentrations of substrate () and product (-----------) are both normalised with respect to [S]". The normalised reactor length (i.e. I = lVmax/F, where Vmax is the maximum velocity for unit reactor length and I is the reactor length) is relative to the length (i.e. when I = 1) that contains sufficient enzyme to convert all the substrate at the given flow rate if the enzyme acted at its maximum velocity throughout; the actual reactor length necessary for complete conversion being longer due to the reduction in the substrate concentration as the reaction progresses. P may be considered as the relative position within a PBR or the reactor's absolute length. ________________________________________ 16.2 Membrane reactors The main requirement for a membrane reactor (MR) is a semipermeable membrane which allows the free passage of the product molecules but contains the enzyme molecules. A cheap example of such a membrane is the dialysis membrane used for
removing low molecular weight species from protein preparations. The usual choice for a membrane reactor is a hollow-fibre reactor consisting of a preformed module containing hundreds of thin tubular fibres each having a diameter of about 200 m and a membrane thickness of about 50 m. Advantage of using Membrane reactor [1] Membrane reactors may be used in either batch or continuous mode and [2] they allow the easy separation of the enzyme from the product. [3] They are normally used with soluble enzymes, avoiding the costs and problems associated with other methods of immobilization and some of the diffusion limitations of immobilized enzymes. If the substrate is able to diffuse through the membrane, it may be introduced to either side of the membrane with respect to the enzyme; otherwise it must be within the same compartment as the enzyme, a configuration that imposes a severe restriction on the flow rate through the reactor, if used in continuous mode.[4] Due to the ease with which membrane reactor systems may be established, they are often used for production on a small scale (g to kg), especially where a multi-enzyme pathway or coenzyme regeneration is needed.[5] They allow the easy replacement of the enzyme in processes involving particularly labile enzymes and can also be used for biphasic reactions. The major disadvantage of these reactors concerns the cost of the membranes and their need to be replaced at regular intervals. 16.3 The kinetics of membrane reactors The kinetics of membrane reactors are similar to those of the batch STR, in batch mode, or the CSTR, in continuous mode (see later). Deviations from these models occur primarily in configurations where the substrate stream is on the side of the membrane opposite to the enzyme and the reaction is severely limited by its diffusion through the membrane and the products' diffusion in the reverse direction. Under these circumstances the reaction may be even more severely affected by product inhibition or the limitations of reversibility than is indicated by these models. 16.4 Packed bed reactors (PBR) The most important characteristic of a PBR is that; they are also called plug flow reactors (PFR) because material flows through the reactor as a plug. Ideally, all of the substrate stream flows at the same velocity, parallel to the reactor axis with no back -mixing. All material present at any given reactor cross -section has had an identical residence time. The longitudinal position within the PBR is, therefore, proportional to the time spent within the reactor; all product emerging with the same residence time and all substrate molecule having an equal opportunity for reaction. The conversion efficiency of a PBR, with respect to its length, behaves in a manner similar to that of a well -stirred batch reactor with respect to its reaction time (Figure 5.2(b)) Each volume element behaves as a batch reactor as it passes through the PBR. Any required degree of reaction may be achieved by use of an idea PBR of suitable length. The flow rate (F) is equivalent to VolS/t for a batch reactor. Therefore equation (16.5) may be converted to represent an ideal PBR, given the assumption, not often realised in practice, that there are no diffusion limitations: (16.6) In order to produce ideal plug -flow within PBRs, a turbulent flow regime is preferred to laminar flow, as this causes improved mixing and heat transfer normal to the flow and reduced axial back-mixing. Achievement of high enough Re may, however, be difficult due to unacceptably high feed rates. Consequent upon the plug -flow characteristic of the PBR is that the substrate concentration is maximised, and the product concentration minimised, relative to the final conversion at every point within the reactor; the effectiveness factor being high on entry to the reactor and low close to the exit. This means that PBRs are the preferred reactors, all other factors being equal, for processes involving product inhibition, substrate activation and reaction reversibility. At low Re the flow rate is proportional to the pressure drop across the PBR. This pressure drop is, in turn, generally found to be proportional to the bed height, the linear flow
rate and dynamic viscosity of the substrate stream and (1 - )2/3 (where is the porosity of the reactor; i.e. the fraction of the PBR volume taken up by the liquid phase), but inversely proportional to the cross-sectional area of the immobilised enzyme pellets. In general PBRs are used with fairly rigid immobilised-enzyme catalysts (1 -3 mm diameter), because excessive increases in this flow rate may distort compressible or physically weak particles. Particle deformation results in reduced catalytic surface area of particles contacting the substrate-containing solution, poor external mass transfer characteristics and a restriction to the flow, causing increased pressure drop. A vicious circle of increased back-pressure, particle deformation and restricted flow may eventually result in no flow at all through the PBR. PBRs behave as deep-bed filters with respect to the substrate stream. It is necessary to use a guard bed if plugging of the reactor by small particles is more rapid than the biocatalysts' deactivation. They are also easily fouled by colloidal or precipitating material. The design of PBRs does not allow for control of pH, by addition of acids or bases, or for easy temperature control where there is excessive heat output, a problem that may be particularly noticeable in wide reactors (> 15 cm diameter). Deviations from ideal plug-flow are due to back-mixing within the reactors, the resulting product streams having a distribution of residence times. In an extreme case, back-mixing may result in the kinetic behaviour of the reactor approximating to that of the CSTR (see below), and the consequent difficulty in achieving a high degree of conversion. These deviations are caused by channeling, where some substrate passes through the reactor more rapidly, and hold-up, which involves stagnant areas with negligible flow rate. Channels may form in the reactor bed due to excessive pressure drop, irregular packing or uneven application of the substrate stream, causing flow rate differences across the bed. The use of a uniformly sized catalyst in a reactor with an upwardly flowing substrate stream reduces the chance and severity of non-ideal behavior. 16.5 Continuous flow stirred tank reactors (CSTIR) This reactor consists of a well -stirred tank containing the enzyme, which is normally immobilised. The substrate stream is continuously pumped into the reactor at the same time as the product stream is removed. This is the example of continous reactor. If the reactor is behaving in an ideal manner, there is total back-mixing and the product stream is identical with the liquid phase within the reactor and invariant with respect to time. Some molecules of substrate may be removed rapidly from the reactor, whereas others may remain for substantial periods. The distribution of residence times for molecules in the substrate stream is shown in Figure 16.2 Advantages The CSTR is an easily constructed, versatile and cheap reactor, which allows simple catalyst charging and replacement. Its well -mixed nature permits straightforward control over the temperature and pH of the reaction and the supply or removal of gases. CSTRs tend to be rather large as the: need to be efficiently mixed. Their volumes are usually about five to ten time the volume of the contained immobilised enzyme. This, however, has the advantage that there is very little resistance to the flow of the substrate stream, which may contain colloidal or insoluble substrates, so long as the insoluble particles are not able to sweep the immobilised enzyme from the reactor. The mechanical nature of the stirring limits the supports for the immobilised enzymes to materials which do not easily disintegrate to give 'fines' which may enter the product stream. However, fairly small particle (down to about 10 m diameter) may be used, if they are sufficiently dense to stay within the reactor. This minimises problems due to diffusional resistance. An ideal CSTR has complete back -mixing resulting in a minimisation of the substrate concentration, and a maximisation of the product concentration, relative
to the final conversion, at every point within the reactor the effectiveness factor being uniform throughout. Thus, CSTRs are the preferred reactors, everything else being equal, for processes involving substrate inhibition or product activation. They are also useful where the substrate stream contains an enzyme inhibitor, as it is diluted within the reactor. This effect is most noticeable if the inhibitor concentration is greater than the inhibition constant and [S]0/Km is low for competitive inhibition or high for uncompetitive inhibition, when the inhibitor dilution has more effect than the substrate dilution. Deviations from ideal CSTR behaviour occur when there is a less effective mixing regime and may generally be overcome by increasing the stirrer speed, decreasing the solution viscosity or biocatalyst concentration or by more effective reactor baffling. Kinetics of CSTR The rate of reaction within a CSTR can be derived from a simple mass balance to be the flow rate (F) times the difference in substrate concentration between the reactor inlet and outlet. Hence: (16.7) Therefore: (16.8) from equation (5.4): (16.9) Therefore: (16.10) This equation should be compared with that for the PBR (equation (16.6)). Together these equations can be used for comparing the productivities of the two reactors (Figures 16.4 and 16.5). ________________________________________ Figure 16. 3Figure 5.4. The residence time distribution of a CSTR. The relative number of molecules resident within the reactor for a particular time N, is plotted against the normalised residence time (i.e. t F/V, where V is the reactor volume, and F is the flow rate; it is the time relative to that required for one reactor volume to pass through the reactor). The residence time distribution of non -reacting media molecules ( ----------- which obeys the relationship , where [M] is the concentration of media molecules, giving a half-life for remaining in the reactor of , product ( ) and substrate () are shown. The reaction S P is assumed, and substrate molecules that have long residence times are converted into product, the average residence time of the product being greater than that for the substrate. The composition of the product stream is identical with that of the liquid phase within the reactor. This composition may be calculated from the relative areas under the curves and, in this case, represents a 90% conversion. Under continuous operating conditions (operating time > 4V/F), the mean residence time within the reactor is V/F. However, it may be noted from the graph that only a few molecules have a residence time close to this value (only 7% between 0.9V/F and 1.1 V/F) whereas 20% of the molecules have residence times of less than 0.1 V/F or greater than 2.3V/F. It should be noted that 100% of the molecules in an equivalent ideal PBR might be expected to have residence times equal to their mean residence time. ________________________________________ Figure 16. 4Figure 5.5. Comparison of the changes in fractional conversion with flow rate between the PBR () and CSTR (-----------) at different values of [S]0/Km (10,1 and 0.1, higher [S]0/Km giving the higher curves). The flow rate is normalised with respect to the reactor's volumetric enzyme content ( = FKm/Vmax. It can be seen that there is little difference between the two reactors at faster flow rates and lower conversions, especially at high values of [S]0/Km. ________________________________________
Figure 16. 5 Figure 5.6. Comparison of the changes in fractional conversion with residence time between the PBR () and CSTR (-----------) at different values of [S]0/Km (10, 1 and 0.1; higher [S]0/Km values giving the lower curves). The residence time is the reciprocal of the normalised flow rate (see Figure 5.5). If the flow rate is unchanged then the 'normalised residence time' may be thought of as the reactor volume needed to produce the required degree of conversion. ________________________________________ Equations describing the behaviour of CSTRs and PBRs utilising reversible reactions or undergoing product or substrate inhibition can be derived in a similar manner, using equations (1.68), (1.85) and (1.96) rather than (1.8): a. Substrate -inhibited PBR (16.11) Substrate -inhibited CSTR (16.12) b. Product -inhibited PBR (16.13) Product -inhibited CSTR (16.14) Reversible reaction in a PBR (16.15) Reversible reaction in a CSTR (16.16) X in equations (5.15) and (5.16) is the fractional conversion for a reversible reaction. (16.17) These equations may be used to compare the size of PBR and CSTR necessary to achieve the same conversion under various conditions (Figure 16.6). The productivity Another useful parameter for comparing these reactors is the productivity. This can be derived for each reactor assuming a first-order inactivation of the enzyme (equation (1.26)). Combined with equation (5.6) for PBR, or (5.10) for CSTR, the following relationships are obtained on integration: PBR (16.18) CSTR (16.19)
where kd is the first -order inactivation constant (i.e. kd1, in equation (1.25)) and the fractional conversion subscripts refer to time = 0 or t. The change in productivity (Figure 16.7) and fractional conversion (Figure 16.8) of these reactors with time can be compared using these equations. These reactors may be operated for considerably longer periods than that determined by the inactivation of their contained immobilised enzyme, particularly if they are capable of high conversion at low substrate concentrations (Figure 16.8). This is independent of any enzyme stabilisation and is simply due to such reactors initially containing large amounts of redundant enzyme. ________________________________________ Figure 16. 6Figure 5.7. Comparison of the ratio, of the enzyme content in a CSTR to that in a PBR, necessary to achieve various degrees of conversion for a range of process conditions. The actual size of the CSTR will be five to ten times greater than indicated due to the necessity of maintaining stirring within the vessel. Uninhibited reaction; product inhibited; --------substrate inhibited. (curve a) [S]0/Km = 100; (curve b) [S]0/Km = 1; (curve c) [S]0/Km < 0.01; (curve d) [S]0/Km = 1, product inhibited KP/Km = 0.1 ; (curve e) [S]0/Km = 1, product inhibited KP/Km = 0.01; (curve f) [S]0/Km = 1, substrate inhibited [S]0/KS = 10; (curve g) [S]0/Km = 100; substrate inhibited [S]0/KS = 10. The size of a CSTR becomes prohibitively large at high conversions (e.g. using
curve b, a CSTR contains three times the enzyme in a PBR to achieve a 90% conversion, but this increases to 18 times for 99% conversion. The difference between the two types of reactor is increased if the effectiveness factor () is less than one due to diffusional effects. ________________________________________ Figure 16. 7Figure 5.8. The change in productivity of a PBR (---------) and CSTR () with time, assuming an initial fractional conversion (X0) = 0.99 and [S]0/Km = 100. The units of time are half -lives of the free enzyme ( ) and the productivity is given in terms of (FXt1/2). Although the overall productivity is 1.6 times greater for a CSTR than a PBR, it should be noted that the CSTR contains 1.9 times more enzyme. ________________________________________ In general, there is little or no back -pressure to increased flow rate through the CSTR. Such reactors may be started up as batch reactors until the required degree of conversion is reached, when the process may be made continuous. CSTRs are not generally used in processes involving high conversions but a chain of CSTRs may approach the PBR performance. This chain may be a number (greater than three) of reactors connected in series or a single vessel divided into compartments, in order to minimise back-mixing CSTRs may be used with soluble rather than immobilised enzyme if an ultrafiltration membrane is used to separate the reactor output stream from the reactor contents. This causes a number of process difficulties, including concentration polarization or inactivation of the enzyme on the membrane but may be preferable in order to achieve a combined reaction and separation process or where a suitable immobilised enzyme is not readily available. ________________________________________ Figure 16. 8Figure 5.9. The change in fractional conversion of PBRs and CSTRs with time, assuming initial fractional conversion (X0) of 0.99 or 0.80. CSTR, [S]o/Km = 0.01; - - - - - - CSTR, [S]0/Km = 100; PBR [S]0/Km = 0.01; ------- PBR [S]0/Km = 100. The time is given in terms of the half -life of the free enzyme ( ). Although the CSTR maintains its fractional conversion for a longer period than the PBR, particularly at high X0. It should be noted that a CSTR capable of X0 = 0.99 at a substrate feed concentration of [S]0/Km = 0.01 contains 22 times more enzyme than an equivalent PBR, but yields only 2.2 times more product. The initial stability in the fractional conversion over a considerable period of time, due to the enzyme redundancy, should not be confused with any effect due to stabilisation of the immobilised enzyme. 5. Fluidised bed reactors These reactors generally behave in a manner intermediate between CSTRs and PBRs. They consist of a bed of immobilised enzyme which is fluidised by the rapid upwards flow of the substrate stream alone or in combination with a gas or secondary liquid stream, either of which may be inert or contain material relevant to the reaction. A gas stream is usually preferred as it does not dilute the product stream. There is a minimum fluidisation velocity needed to achieve bed expansion, which depends upon the size, shape, porosity and density of the particles and the density and viscosity of the liquid. This minimum fluidisation velocity is generally fairly low (about 0.2 -I.0 cm s-1) as most immobilisedenzyme particles have densities close to that of the bulk liquid. In this case the relative bed expansion is proportional to the superficial gas velocity and inversely proportional to the square root of the reactor diameter. Fluidising the bed requires a large power input but, once fluidised, there is little further energetic input needed to increase the flow rate of the substrate stream through the reactor (Figure 16.2). At high flow rates and low reactor diameters almost ideal plug -flow characteristics may be achieved. However, the kinetic performance of the FBR normally lies between that of the PBR and the CSTR, as the small fluid linear velocities allowed by most biocatalytic particles causes a degree of back-
mixing that is often substantial, although never total. The actual design of the FBR will determine whether it behaves in a manner that is closer to that of a PBR or CSTR (see Figures 5.5 -5.9). It can, for example, be made to behave in a manner very similar to that of a PBR, if it is baffled in such a way that substantial backmixing is avoided. FBRs are chosen when these intermediate characteristics are required, e.g. where a high conversion is needed but the substrate stream is colloidal or the reaction produces a substantial pH change or heat output. They are particularly useful if the reaction involves the utilisation or release of gaseous material. The FBR is normally used with fairly small immobilised enzyme particles (20-40 m diameter) in order to achieve a high catalytic surface area. These particles must be sufficiently dense, relative to the substrate stream, that they are not swept out of the reactor. Less-dense particles must be somewhat larger. For efficient operation the particles should be of nearly uniform size otherwise a non-uniform biocatalytic concentration gradient will be formed up the reactor. FBRs are usually tapered outwards at the exit to allow for a wide range of flow rates. Very high flow rates are avoided as they cause channelling and catalyst loss. The major disadvantage of development of FBR process is the difficulty in scalingup these reactors. PBRs allow scale-up factors of greater than 50000 but, because of the markedly different fluidisation characteristics of different sized reactors, FBRs can only be scaled-up by a factor of 10 -100 each time. In addition, changes in the flow rate of the substrate stream causes complex changes in the flow pattern within these reactors that may have consequent unexpected effects upon the conversion rate.
Chapter-17
Protein Engineering
Introduction Protein engineering is the branch of science in which novel enzymes are being designed and produce by genetic engineering methods to improve the stability of an enzyme (stability with respect to chemical oxidation, temperature, and pH). Therefore protein engineering includes two parts one large scale production of genes by modification in the gene by gene cloning methods and other modification in the structure of protein to improve the substrate specificity, the cofactor requirement (NADPH to NADH) or imparting novel properties to a protein (eg. adding a Mn binding site into horse heart myoglobin at UBC).In the genetic engineering methods the availability of a cloned gene is important. Cloned gene is expressed in the expression vector to obtain the 3-D structure of the enzyme, resolved by Xray crystallography. In the absence of detailed 3-D structure, one can do random mutagenesis and/or directed evolution. There are many directions in which enzyme technologists are currently applying their art and which are at the forefront of biotechnological research and development. At present, relatively few enzymes are available on a large scale (i.e. > kg) and are suitable for industrial applications. These shortcomings are being addressed in a number of ways: New enzymes are being sought in the natural environment and by strain selection. Established industrial enzymes are being used in as wide a variety of ways as can be conceived. novel enzymes are being designed and produce by genetic engineering;
New organic catalysts are being designed and synthesized. More complex enzyme systems are being utilised. The development of genetically improved enzymes is generally undertaken by molecular biologists and the design and synthesis of novel enzyme-like catalysts is done by the organic chemists. Both groups of workers will, however, base their science on data provided by the enzyme technologist. Space requirements in this volume do not allow the full treatment of these related areas but will be discussed briefly here. 17.1 Enzyme engineering A most exciting development over the last few years is the application of genetic engineering techniques to enzyme technology. There are a number of properties which may be improved or altered by genetic engineering for example, the yield and kinetics of the enzyme, the ease of downstream processing and various safety aspects. Enzymes from dangerous or unapproved microorganisms and from slow growing or limited plant or animal tissue may be cloned into safe high-production microorganisms. In the future, enzymes may be redesigned to fit more appropriately into industrial processes; for example, making glucose isomerase less susceptible to inhibition by the Ca2+ present in the starch saccharification processing stream. 17.2 Enzyme production can be increased by cloning of gene The amount of enzyme produced by a microorganism may be increased by increasing the number of gene copies that code for it by inserting a portion of gene for specific enzyme into the suitable vector. If the vector is expression type then large amount of protein will be obtained after the translation of vector. The product can be tested by its specific function. The DNA having gene of interest for specific enzyme can be isolated by partial digestion of gene or complete digestion of gene (incomplete or complete fragmentation of gene) and selection of specific gene can be done with the help of probe. This principle has been used to increase the activity of penicillin-G-amidase in Escherichia coli. The cellular DNA from a producing strain is selectively cleaved by the restriction endonuclease Hind III. This hydrolyses the DNA at relatively rare sites containing the 5'-AAGCTT-3' base sequence to give identical 'staggered' ends. Figure 17. 1 figure showing staggered cut of DNA double strand by the restriction endonuclease. Note that single stranded DNA is rarely cut by the Restriction enzyme.
R.E. Intact DNA
cleaved DNA
Figure 17. 2 showing structure of vector and their site containing antibiotic resistance gene as the marker of the gene for selection of recombinant gene.
The total DNA is cleaved into about 10000 fragments, only one of which contains the required genetic information. These fragments are individual cloned into a cosmid vector and thereby returned to E. coli. These colonies containing the active gene are identified by their inhibition of a 6-amino-penicillanic acidsensitive organism. Such colonies are isolated and the penicillin-G-amidase gene transferred on to pBR322 plasmids and recloned back into E. coli. The engineered cells, aided by the plasmid amplification at around 50 copies per cell, produce penicillin-G-amidase constitutively and in considerably higher quantities than does the fully induced parental strain. Such increased yields are economically relevant not just for the increased volumetric productivity but also because of
reduced downstream processing costs, the resulting crude enzyme being that much purer. Another extremely promising area of genetic engineering is protein engineering. New enzyme structures may be designed and produced in order to improve on existing enzymes or create new activities. An outline of the process of protein engineering is shown in Figure 17.1. Such factitious enzymes are produced by site-directed mutagenesis (Figure 17.2). Unfortunately from a practical point of view, much of the research effort in protein engineering has gone into studies concerning the structure and activity of enzymes chosen for their theoretical importance or ease of preparation rather than industrial relevance. This emphasis is likely to change in the future. Figure 17. 3 showing structure and location of plasmid DNA inside the E.coli vector. Figure 17. 4 showing nature of cutting of gene one sticky end Figure 17. 5 showing selection of recombinant by use antibiotic
As indicated by the method used for site-directed mutagenesis , the preferred pathway for creating new enzymes is by the stepwise substitution of only one or two amino acid residues out of the total protein structure. Although a large database of sequence-structure correlations is available, (NCBI) and growing rapidly together with the necessary software, it is presently insufficient accurately to predict three-dimensional changes as a result of such substitutions. The main problem is assessing the long-range effects, including solvent interactions, on the new structure. As the many reported results would attest, the science is at a stage where it can explain the structural consequences of amino acid substitutions after they have been determined but cannot accurately predict them.
Figure 17. 6 showing complete procedure of gene cloning.
Figure 17. 7 The protein engineering cycle. The process starts with the isolation and characterization of the required enzyme. This information is analyzed together with the database of known and putative structural effects of amino acid substitutions to produce a possible improved structure. This factitious enzyme is constructed by site-directed mutagenesis, isolated and characterised. The results, successful or unsuccessful, are added to the database, and the process repeated until the required result is obtained. ________________________________________ Protein engineering, therefore, is presently rather a hit or miss process which may be used with only little realistic likelihood of immediate success. Apparently quite small sequence changes may give rise to large conformational alterations and even affect the rate-determining step in the enzymic catalysis. However it is reasonable to suppose that, given a sufficiently detailed database plus suitable software, the relative probability of success will increase over the coming years and the products of protein engineering will make a major impact on enzyme technology. 17.2 Application of Enzyme Engineering 17.2.1 Subtilisin Much protein engineering has been directed at subtilisin (from Bacillus amyloliquefaciens), the principal enzyme in the detergent enzyme preparation,
Alcalase. This has been aimed at the improvement of its activity in detergents by stabilising it at even higher temperatures, pH and oxidant strength. Most of the attempted improvements have concerned alterations to: The P1 cleft, which holds the amino acid on the carbonyl side of the targeted peptide bond; 2. The oxyanion hole (principally Asn155), which stabilises the tetrahedral intermediate; 3. the neighborhood of the catalytic histidyl residue (His64), which has a general base role; and 4.the methionine residue (Met222) which causes subtilisin's lability to oxidation. It has been found that the effect of a substitution in the P1 cleft on the relative specific activity between substrates may be fairly accurately predicted even though predictions of the absolute effects of such changes are less successful. Many substitutions, particularly for the glycine residue at the bottom of the P1 cleft (Gly166), have been found to increase the specificity of the enzyme for particular peptide links whilst reducing it for others. These effects are achieved mainly by corresponding changes in the Km rather than the Vmax. Increases in relative specificity may be useful for some applications. They should not be thought of as the usual result of engineering enzymes, however, as native subtilisin is unusual in being fairly non-specific in its actions, possessing a large hydrophobic binding site which may be made more specific relatively easily (e.g. by reducing its size). The inactivation of subtilisin in bleaching solutions coincides with the conversion of Met222 to its sulfoxide, the consequential increase in volume occluding the oxyanion hole. Substitution of this methionine by serine or alanine produces mutants that are relatively stable, although possessing somewhat reduced activity. ________________________________________ Figure 17. 8 Figure 17. 9 An outline of the process of site-directed mutagenesis, using a hypothetical example. (a) The primary structure of the enzyme is derived from the DNA sequence. A putative enzyme primary structure is proposed with an asparagine residue replacing the serine present in the native enzyme. A short piece of DNA (the primer), complementary to a section of the gene apart from the base mismatch, is synthesised. (b) The oligonucleotide primer is annealed to a single-stranded copy of the gene and is extended with enzymes and nucleotide triphosphates to give a double-stranded gene. On reproduction, the gene gives rise to both mutant and wild-type clones. The mutant DNA may be identified by hybridisation with radioactively labelled oligonucleotides of complementary structure. ________________________________________ Trypsin An example of the unpredictable nature of protein engineering is given by trypsin, which has an active site closely related to that of subtilisin. Substitution of the negatively charged aspartic acid residue at the bottom of its P1 cleft (Asp189), which is used for binding the basic side-chains of lysine or arginine, by positively charged lysine gives the predictable result of abolishing the activity against its normal substrates but unpredictably also gives no activity against substrates where these basic residues are replaced by aspartic acid or glutamic acid. Thermophilic enzymes Considerable effort has been spent on engineering more thermophilic enzymes. It has been found that thermophilic enzymes are generally only 20-30 kJ more stable than their mesophilic counterparts. This may be achieved by the addition of just a few extra hydrogen bonds, an internal salt link or extra internal hydrophobic residues, giving a slightly more hydrophobic core. All of these changes are small enough to be achieved by protein engineering. To ensure a more predictable outcome, the secondary structure of the enzyme must be conserved and this
generally restricts changes in the exterior surface of the enzyme. Suitable for exterior substitutions for increasing thermostability have been found to be aspartate < glutamate lysine, glutamine, valine < threonine, serine < asparagine, isoleucine < threonine, asparagine <aspartate and lysine < arginine. Such substitutions have a fair probability of success. Whenever there is small increases in the interior hydrophobicity, for example by substituting interior glycine or serine residues by alanine may also increase the thermostability. It should be recognized that making an enzyme more thermostable reduces its overall flexibility and, hence, it is probable that the factitious enzyme produced will have reduced catalytic efficiency. Artificial enzymes Synzymes A number of possibilities now exist for the construction of artificial enzymes. These are generally synthetic polymers or oligomers with enzyme-like activities, often called synzymes. They must possess two structural entities, a substratebinding site and a catalytically effective site. It has been found that producing the facility for substrate binding is relatively straightforward but catalytic sites are somewhat more difficult. Both sites may be designed separately but it appears that, if the synzyme has a binding site for the reaction transition state, this often achieves both functions. Synzymes generally obey the saturation Michaelis-Menten kinetics. For a one-substrate reaction the reaction sequence is given by synzyme + S (synzyme-S complex) synzyme + P [17.5] Some synzymes are simply derivatised proteins, although covalently immobilised enzymes are not considered here. An example is the derivatisation of myoglobin, the oxygen carrier in muscle, by attaching (Ru(NH3)5)3+ to three surface histidine residues. This converts it from an oxygen carrier to an oxidase, oxidising ascorbic acid whilst reducing molecular oxygen. The synzyme is almost as effective as natural ascorbate oxidases. Disadvantage of synzymes It is impossible to design protein synzymes from scratch with any probability of success, as their conformations are not presently predictable from their primary structure. Such proteins will also show the drawbacks of natural enzymes, being sensitive to denaturation, oxidation and hydrolysis. For example, polylysine binds anionic dyes but only 10% as strongly as the natural binding protein, serum albumin, in spite of the many charges and apolar side-chains. Polyglutamic acid, however, shows synzymic properties. It acts as an esterase in much the same fashion as the acid proteases, showing a bell-shaped pH-activity relationship, with optimum activity at about pH 5.3, and Michaelis-Menten kinetics with a Km of 2 mm and Vmax of 10-4 to 10-5 s-1 for the hydrolysis of 4-nitrophenyl acetate. Cyclodextrins (Schardinger dextrins) are naturally occurring toroidal molecules consisting of six, seven, eight, nine or ten -1, 4-linked D-glucose units joined head-to-tail in a ring and -cyclodextrins, respectively: they may be synthesized from starch by the cyclomaltodextrin glucanotransferase (EC 2.4.1.19) from Bacillus macerans). They differ in the diameter of their cavities (about 0.5-1 nm) but all are about 0.7 nm deep. These form hydrophobic pockets due to the glycosidic oxygen atoms and inwards-facing C-H groups. All the C-6 hydroxyl groups project to one end and all the C-2 and C-3 hydroxyl groups to the other. Their overall characteristic is hydrophilic, being water soluble, but the presence of their hydrophobic pocket enables them to bind hydrophobic molecules of the appropriate size. Synzymic cyclodextrins are usually derivatised in order to introduce catalytically relevant groups. Many such derivatives have been examined. For example, a C-6 hydroxyl group of -cyclodextrin was covalently derivatised by an activated pyridoxal coenzyme. The resulting synzyme not only acted a transaminase (see reaction scheme [1.2]) but also showed stereoselectivity for the L-amino acids. It was not as active as natural transaminases, however. Polyethylenimine is formed by polymerising ethyleneimine to give a highly branched
hydrophilic three-dimensional matrix. About 25% of the resultant amines are primary, 50% secondary and 25% tertiary: [17.6] Ethyleneimine polyethyleneimine The primary amines may be alkylated to form a number of derivatives. If 40% of them are alkylated with 1-iodododecane to give hydrophobic binding sites and the remainder alkylated with 4(5)-chloromethylimidazole to give general acid-base catalytic sites, the resultant synzyme has 27% of the activity of -chymotrypsin against 4-nitrophenyl esters. As might be expected from its apparently random structure, it has very low esterase specificity. Other synzymes may be created in a similar manner. Antibodies to transition state analogues of the required reaction may act as synzymes. For example, phosphonate esters of general formula (R-PO2-OR')- are stable analogues of the transition state occurring in carboxylic ester hydrolysis. Monoclonal antibodies raised to immunizing protein conjugates covalently attached to these phosphonate esters act as esterases. The specificities of these catalytic antibodies (also called abzymes) depends on the structure of the side-chains (i.e. R and R' in (R-PO2-OR')-) of the antigens. The Km values may be quite low, often in the micromolar region, whereas the Vmax values are low (below 1 s-1), although still 1000-fold higher than hydrolysis by background hydroxyl ions. A similar strategy may be used to produce synzymes by molecular 'imprinting' of polymers, using the presence of transition state analogues to shape polymerising resins or inactive non-enzymic protein during heat denaturation. 17.3 Advantages of genetic engineering Many enzymes are now produced by fermentation of genetically modified microrganisms (GMOs). There are several advantages of using GMOs for the production of enzymes, including. It is possible to produce enzymes with a higher specificity and purity. It is possible to obtain enzymes which would otherwise not be available for economical, occupational health or environmental reasons. Due to higher production efficiency there is an additional environmental benefit through reducing energy consumption and waste from the production plants For enzymes used in the food industry particular benefits are for example a better use of raw materials (juice industry), better keeping quality of a final food and thereby less wastage of food (baking industry) and a reduced use of chemicals in the production process (starch industry). For enzymes used in the feed industry particular benefits include a significant reduction in the amount of phosphorus released to the environment from farming. The enzymes are produced by fermentation of the genetically modified microrganisms (the production strain), which then produces the desired enzyme. The process takes place under well controlled conditions in closed fermentation tank. After fermentation the enzyme is separated from the production strain, purified and mixed with inert diluents for stabilisation. Genetic engineering can be used to improve production and purification of recombinant enzymes, eg. targeting enzymes for extracellular secretion to allow for their easy isolation and purification. Examples of industrially useful enzymes include: A. proteases: subtilisin (detergent enzyme), rennet (cheese-processing). B. carbohydrases: amylases (starch hydrolysis), cellulases (bioconversions by Stake Technology), xylanases (bioconversions and biopulping), glucose isomerase (HFCS) C. pharmaceutically important enzymes: urokinase (plasminogen activator) is a protease for the treatment of thrombosis and embolism (Prve et al. 1987). The Lilly Research Labs hold the patent on the production of urokinase from a continuous line of porcine kidney cells D. ligninases and Mn peroxidases: pulp processing.
E. restriction endonucleases: used extensively in research.
Chapter-18
Biosensors and Immunosensors
Introduction A biosensor is an analytical device which converts a biological response into an electrical signal (Figure 19.1). The term 'biosensor' is often used to cover sensor devices used in order to determine the concentration of substances. Clark and Lyons first demonstrated the modern concept of biosensors, in which an enzyme was immobilized into an electrode to form a biosensor. When the biological component of biosensor is a component of the immune system then it is referred to as Immunosensors. The emphasis of this Chapter concerns enzymes as the biologically responsive material, but it should be recognised that other biological systems may be utilised by biosensors, for example, whole cell metabolism, ligand binding and the antibody-antigen reaction. 19.1 The use of enzymes in analysis Enzymes make excellent analytical reagents due to their specificity, selectivity and efficiency. They are often used to determine the concentration of their substrates (as analytes) by means of the resultant initial reaction rates. If the reaction conditions and enzyme concentrations are kept constant, these rates of reaction (v) are proportional to the substrate concentrations ([S]) at low substrate concentrations. When [S] < 0.1 Km, equation from Michaelis velocity constant simplifies to give v = (Vmax/Km)[S] The rates of reaction are commonly determined from the difference in optical absorbance between the reactants and products. An example of this is the -Dgalactose dehydrogenase (EC 1.1.1.48) assay for galactose which involves the oxidation of galactose by the redox coenzyme, nicotine-adenine dinucleotide (NAD+). -D-galactose + NAD+ D-galactono-1,4-lactone + NADH + H+ [19.1] A 0.1 mM solution of NADH has an absorbance at 340nm, in a 1 cm path-length cuvette, of 0.622, whereas the NAD+ from which it is derived has effectively zero absorbance at this wavelength. The conversion (NAD+ NADH) is, therefore, accompanied by a large increase in absorption of light at this wavelength. For the reaction to be linear with respect to the galactose concentration, the galactose is kept within a concentration range well below the Km of the enzyme for galactose. In contrast, the NAD+ concentration is kept within a concentration range well above the Km of the enzyme for NAD+, in order to avoid limiting the reaction rate. Such assays are commonly used in analytical laboratories and are, indeed, excellent where a wide variety of analyses need to be undertaken on a relatively small number of samples. The drawbacks to this type of analysis become apparent when a large number of repetitive assays need to be performed. Then, they are seen to be costly in terms of expensive enzyme and coenzyme usage, time consuming, labour intensive and in need of skilled and reproducible operation within properly equipped analytical laboratories. For routine or on-site operation, these disadvantages must be overcome. This is being achieved by the production of biosensors which exploit biological systems in association with advances in micro-electronic technology. Research and development in this field is wide and multidisciplinary, spanning biochemistry, bioreactor science, physical chemistry, electrochemistry, electronics and software engineering. Most of this current endeavour concerns potentiometric and amperometric biosensors and colorimetric paper enzyme strips. 19.2 A successful biosensor must possess at least some of the following beneficial features
The biocatalyst must be highly specific for the purpose of the analyses, be stable under normal storage conditions and, except in the case of colorimetric enzyme strips and dipsticks (see later), show good stability over a large number of assays (i.e. much greater than 100). The reaction should be as independent of such physical parameters as stirring, pH and temperature as is manageable. This would allow the analysis of samples with minimal pre-treatment. If the reaction involves cofactors or coenzymes these should, preferably, also be co-immobilised with the enzyme. The response should be accurate, precise, reproducible and linear over the useful analytical range, without dilution or concentration. It should also be free from electrical noise. If the biosensor is to be used for invasive monitoring in clinical situations, the probe must be tiny and biocompatible, having no toxic or antigenic effects. If it is to be used in fermentors it should be sterilisable. This is preferably performed by autoclaving but no biosensor enzymes can presently withstand such drastic wet-heat treatment. In either case, the biosensor should not be prone to fouling or proteolysis. The complete biosensor should be cheap, small, portable and capable of being used by semi-skilled operators. There should be a market for the biosensor. There is clearly little purpose developing a biosensor if other factors (e.g. government subsidies, the continued employment of skilled analysts, or poor customer perception) encourage the use of traditional methods and discourage the decentralisation of laboratory testing. 19.2.2 Some of the notable characteristics of biosensor Specificity: Biosensor has got the remarkable ability to distinguish between the analyte of interest and other similar substances. With biosensor, an analyte can be detected with great accuracy . Response time: Analytic tracers or catalytic product can be detected directly and instantaneously with the help of biosensors. Thus they are very much suitable for on-line monitoring. Simplicity: The sensing molecule and the transducer are integrated on to a single probe. Thus handling of biosensors is very easy. Continuous monitoring ability: Biosensors can be regenerated and reused. Thus they are suitable for continuous monitoringJ purposes which can never be done with conventional analysis methods. Reproducibility: Biosensors are very reliable; reproducibility of the values is also a great advantage. Portability: Portability of such sensors adds to the advantages which can never be the case with a spectrophotometer. Cost: The cost of the biosensor has too some extend limited its application. 19.3 Working of Biosensor Mechanism of Biosensor ________________________________________ Figure 19. 1 Schematic diagram showing the main components of a biosensor. The biocatalyst (a) converts the substrate to product. This reaction is determined by the transducer (b) which converts it to an electrical signal. The output from the transducer is amplified (c), processed (d) and displayed (e). ________________________________________ A biosensor is a device that detects, transmits and records information regarding a physiological or biochemical change. Technically, it is a probe that integrates a biological component with an electronic transducer thereby converting a biochemical signal into a quantifiable electrical response. The sensor works by converting the signal produced by the biological sensing element on response to a specific analyte to a measurable electrical signal with the help of the transducer. The amplifier increases the intensity of the signal so that it can be
readily measured. The digital display then displays the reading in a suitable unit. All these components are generally integrated onto a single probe to make the handling easier. Biosensor is device that on matching with appropriate biological sample gives signal to electrical components. Biological component is bound or immobilized on the electronic component called as transducer. Figure 19. 2 The electrical signal from the transducer is often low and superimposed upon a relatively high and noisy (i.e. containing a high frequency signal component of an apparently random nature, due to electrical interference or generated within the electronic components of the transducer) baseline. The signal processing normally involves subtracting a 'reference' baseline signal, derived from a similar transducer without any biocatalytic membrane, from the sample signal, amplifying the resultant signal difference and electronically filtering (smoothing) out the unwanted signal noise. The relatively slow nature of the biosensor response considerably eases the problem of electrical noise filtration. The analogue signal produced at this stage may be output directly but is usually converted to a digital signal and passed to a microprocessor stage where the data is processed, converted to concentration units and output to a display device or data store. 19.4 Classification of Biosensor There are several type of Biosensor depends on the physical characteristic like heat , electrical signal . light, electron ,charge, and mass. Biosensors may be classified according to several criteria, such as transducers, bioactive components, or immobilization techniques used. The heat output (or absorbed) by the reaction (calorimetric biosensors), changes in the distribution of charges causing an electrical potential to be produced (potentiometric biosensors), movement of electrons produced in a redox reaction (amperometric biosensors), light output during the reaction or a light absorbance difference between the reactants and products (optical biosensors), or Effects due to the mass of the reactants or products (piezo-electric biosensors). There are three so-called 'generations' of biosensors First generation biosensors where the normal product of the reaction diffuses to the transducer and causes the electrical response. Second generation biosensors which involve specific 'mediators' between the reaction and the transducer in order to generate improved response. Third generation biosensors where the reaction itself causes the response and no product or mediator diffusion is directly involved. A typical biosensor has got two main parts Biological component which can be enzyme, antibody, nucleic acid, microorganism, tissue, cell, etc. Electronic device, i.e., transducer which can be electrochemical, optical, piezoelectric, thermal, etc. The biological component is generally immobilized near or onto the transducer surface in order to facilitate reuse, to minimize interference and to maximize response. In each case it is the ability of the biological component to react or respond specifically to an analyte (or a group of analytes) that makes the biomolecule suitable for use as the sensing element of a biosensor. For example, an antibody will only bind to the specific antigen under suitable conditions. The biological signal that is produced is converted by means of a suitable transducer into a quantifiable electrical signal.Biosensors are suitable for detecting a wide variety of analytes including pollutants, explosives, viruses, biochemical & pharmaceutical products, vitamins, amino acids, heavy metals, ions, gases etc . In fact, every single analyte, be it simple or complex can be detected provided a biomolecule that response specifically to it is identified. 1. Transducers
This is the component of biosensor which converts the biological signal to a quantifiable electrical signal. Electrochemical: In this configuration, sensing molecules are either coated onto or covalently bonded to a probe surface. A membrane holds the sensing molecule in place, excluding interfering species from the analyte solution. The sensing molecule reacts specifically with compounds to be detected, sparking an electrical signal proportional to the concentration of the analyte. The most common detection method for electrochemical biosensors involves measurement of current, voltage, capacitance, conductance and impedance. Among such sensors, amperometric (e.g. Oxygen or hydrogen peroxide) and potentiometric (e.g. pH and carbon dioxide) transducers have found the widest applications. Optical: In optical biosensors, the optical fibers allow detection of analytes on the basis of absorption, fluorescence or light scattering. Since they are nonelectrical, optical biosensors have the advantages of lending themselves to in vivo applications and allowing multiple analytes to be detected by using different monitoring wave-lengths. The versatility of fiber optics probes is due to their capacity to transmit signals that reports on changes in wavelength, wave propagation, time, intensity, distribution of the spectrum or polarity of light. Piezoelectric: In this mode, sensing molecules are attached to a piezoelectric surface-a mass to frequency transducer-in which interactions between the analyte and the sensing molecules set up mechanical vibrations that can be translated into an electrical signal proportional to the analyte. Example of such sensor is quartz crystals. Thermal: In this mode, the biocomponent is immobilized in proximity to the heat sensing transducer, generally a thermister. Most of the enzymatic or microbial reactions are accompanied by considerable heat evolution making this sensor applicable to a very wide range of detection. However the use of sophisticated and expensive instrumentation is the major drawback of this technique. 2. Bioactive components This is the biological part of the biosensor which specifically reacts with the analyte of interest sparking a signal that is detectable by the attached transducer. Enzymes: Purified enzymes have been commonly used in the construction of biosensors due to their high specific activities as well as high analytical specificity. They are mostly used in catalytic type biosensors. Purified enzymes, however, are expensive and unstable, thus limiting their application sin the field of biosensors. As most of the enzymes being employed are intracellular, isolation and purification becomes tough. Antibodies: The binding between an antigen and its corresponding antibody is very specific. This property of antibody is exploited while designing biosensors based on antibodies. The binding reaction between the antibody and antigen can be monitored as a time dependent change of fluorescence signal which is proportional to the reaction ratio of antibody to analyte. Cells: Either whole microorganisms or tissues can be used as the biocomponent. Whole cells can be used either in a viable or non-viable form. Viable microbes metabolize various organic compounds either anaerobically or aerobically resulting in various end products like ammonia, carbon dioxide, acids etc that can be monitored using a variety of transducers. Viable cells are mainly used when the overall substrate assimilation capacity of microorganism is taken as an index of respiratory metabolic activity, as in the case of estimation of BOD, vitamins, sugars, organic acids, etc. Another mechanism used for the viable microbial biosensor involves the inhibition of microbial respiration by the analyte of interest, like environmental pollutants. The major limitation to the use of whole cells is the diffusion of substrate and products through the cell wall resulting in a slow response as compared to enzyme-based sensors. Nucleic acids: The ability of a single stranded nucleic acid to hybridize with another fragment of DNA by complementary base pairing is the principle behind the
nucleic acid sensors. Technological innovation is introduced in the manner in which the nucleic acid oligomer is attached to the surface of the detector and the manner in which the hybridized nucleic acid is detected and transduced into a measurable signal. Ammonia derivetised oligonucleotides can be detected by attaching to glass surfaces such as fiber-optics cables, glass beads or microscopic slides through covalent bonding with a chemical linker. Lipids: An active biological receptor can be immobilized and stabilized in a polymeric film for determining an analyte of interest in a sample. 3. Immobilization techniques for the bio-component: The biological material should bring the physico-chemical changes in close proximity of a transducer. Immobilization not only helps in forming the required close proximity between the biomaterial and the transducer, but also helps in stabilizing it for reuse.The selection of a technique and/or support material would depend on the nature of the biomaterial and the substrate and configuration of the transducer used. Covalent binding, a commonly used technique for the immobilization of enzymes and antibodies, has not been useful for the immobilization of cells. On the other hand, cross-linking using bifunctional reagents like glutaraldehyde has been successfully used for the immobilization of cells in various supports. Thus crosslinking technique will be useful in obtaining immobilized non-viable cell preparations containing active intracellular enzymes. Entrapment and adsorption techniques are more useful when viable cells are used. The synthetic polymers used for microbial biosensor applications include polyacrylamide, polyurethane-based hydrogels, photo cross-linkable resins and polyvinyl alcohol. Natural polymers used for the entrapment of the cells include alginate, carrageenam, low-melting agarose, chitosan, etc. But entrapment technique adds another diffusional barrier.Passive trapping of cells into the pores or adhesion onto the surfaces of cellulose or other synthetic membranes has the major advantage of direct contact between the liquid phase and the cell, thus reducing or eliminating the problem of mass transfer Table 19.1 table gives detail account of different type of Biosensor. Analyte Microorganism Transducer/immobilization Detection limit BOD Trichosporum cutaneum Miniature O2 electrode 0.2-28 mg/L BOD Activated sludge (mixed microbial consortium) O2 electrode/flow injection system >3.5 mg/L Phenolic compounds Ps. Putida O2 electrode (reactor with cells adsorbed on PEI glass) 100 M Nitrite Nitrobactor vulgaris O2 electrode (adsorption on Whatman paper) >10 M Cyanide S. cerevisiae O2 electrode (PVA) 0.15-15 nM Organophosphate nerve agent GEM E.coli Potentiometric 0.055-1.8 mM Mercuric chloride Synechococcus sp. PCC 7942 Photoelectrochemical 0.2 and 0.06 M Alcohol Candida vini O2 electrode (Porous acetyl cellulose filter) 0.20.02 mM Glucose A. niger O2 electrode (entrapment in dialysis membrane) >1.75 mM Glucose, sucrose, lactose G. oxydans, S. cerevisiae, K. marxianus O2 electrode (gelatine) Up to 0-0.8 mM Sugars (glucose) Psychophilic D.radiodurans O2 electrode (agarose) 0.03-0.55 mM Short chain fatty acids in milk A. nicotianae O2 electrode (Polyvinyl alcohol) 0.11-1.7 mM CO2 CO2 utilizing autotrophic bacteria O2 electrode (bound on cellulose nitrate membrane) 0.2-5 mM
Vitamin B-6 S. uvarum O2 electrode (adsorption on cellulose nitrate membrane) 0.5-2.5 ng/ml Vitamin B-12 E. coli O2 electrode (trapped in porous acetyl cellulose membrane) 5-25x10-9 mM Peptides B. subtilis O2 electrode (filter paper strip & dialysis membrane)0.070.6 mM Phenylalanine P. vulgaris Amperometric O2 electrode 2.5x10-2 -2.5mM Pyruvate Streptococcus faecium CO2 gas sensing electrode 0.22-32 mM Tyrosine A.phenologenes NH3 gas sensing electrode 8.2x10-2 -1.0 mM Monitoring toxicity of compounds to eukaryotes S. cerevisiae was genetically modified to express firefly luciferase On-line monitoring of microbial growth E. coli engineered for constitutive bioluminescence Toxicity of Zn, Cu and Cd, alone or in combination E. coli HB101 and Ps. fluorescens 10586 genetically modified with luxCDABE Polycyclic aromatic hydrocarbons Ps. fluorescens HK44 genetically modified with luxCDABE Ecotoxicity assessment of organotins and their initial breakdown products Microtox and luxCDABE modified Ps. fluorescens Ethanol as a model toxicant E. coli TV1061, harboring the plasmid pGrpELux5 Monitoring of biocides Bioluminescent strain of E. coli produced by rDNA technology Metals, solvents, crop protection chemicals E. coli heat shock promoters, dnaK & grpE were fused with lux gene of V. fischeri Assessment of the toxicity of metals in soils amended with sewage sludge luxCDABE modified Ps. fluorescens Ps. fluorescens 19.5Type of Biosensor 19.5.1 Optical biosensors There are two main areas of development in optical biosensors. These involve determining changes in light absorption between the reactants and products of a reaction, or measuring the light output by a luminescent process. The former usually involve the widely established, if rather low technology, use of colorimetric test strips. These are disposable single-use cellulose pads impregnated with enzyme and reagents. The most common use of this technology is for whole-blood monitoring in diabetes control. In this case, the strips include glucose oxidase, horseradish peroxidase (EC 1.11.1.7) and a chromogen (e.g. otoluidine or 3,3',5,5'-tetramethylbenzidine). The hydrogen peroxide, produced by the aerobic oxidation of glucose, oxidising the weakly coloured chromogen to a highly coloured dye. peroxidase chromogen(2H) + H2O2 dye + 2H2O [19.2] The evaluation of the dyed strips is best achieved by the use of portable reflectance meters, although direct visual comparison with a coloured chart is often used. A wide variety of test strips involving other enzymes are commercially available at the present time.A most promising biosensor involving luminescence uses firefly luciferase (Photinus-luciferin 4-monooxygenase (ATP-hydrolysing), EC 1.13.12.7) to detect the presence of bacteria in food or clinical samples. Bacteria are specifically lysed and the ATP released (roughly proportional to the number of bacteria present) reacted with D-luciferin and oxygen in a reaction which produces yellow light in high quantum yield. luciferase ATP + D-luciferin + O2 oxyluciferin + AMP + pyrophosphate + CO2 + light (562 nm) [19.3] The light produced may be detected photometrically by use of high-voltage, and expensive, photomultiplier tubes or low-voltage cheap photodiode systems. The sensitivity of the photomultiplier-containing systems is, at present, somewhat
greater (< 104 cells ml-1, < 10-12 M ATP) than the simpler photon detectors which use photodiodes. Firefly luciferase is a very expensive enzyme, only obtainable from the tails of wild fireflies. Use of immobilised luciferase greatly reduces the cost of these analyses. 19.5.2 Amperometric biosensors Amperometric biosensors function by the production of a current when a potential is applied between two electrodes. They generally have response times, dynamic ranges and sensitivities similar to the potentiometric biosensors. The simplest amperometric biosensors in common usage involve the Clark oxygen electrode (Figure 19.3). This consists of a platinum cathode at which oxygen is reduced and a silver/silver chloride reference electrode. When a potential of -0.6 V, relative to the Ag/AgCl electrode is applied to the platinum cathode, a current proportional to the oxygen concentration is produced. Normally both electrodes are bathed in a solution of saturated potassium chloride and separated from the bulk solution by an oxygen-permeable plastic membrane (e.g. Teflon, polytetrafluoroethylene). The following reactions occur: Ag anode 4Ag0 + 4Cl- 4AgCl + 4e[19.4] Pt cathode O2 + 4H+ + 4e- 2H2O [19.5] The efficient reduction of oxygen at the surface of the cathode causes the oxygen concentration there to be effectively zero. The rate of this electrochemical reduction therefore depends on the rate of diffusion of the oxygen from the bulk solution, which is dependent on the concentration gradient and hence the bulk oxygen concentration. It is clear that a small, but significant, proportion of the oxygen present in the bulk is consumed by this process; the oxygen electrode measuring the rate of a process which is far from equilibrium, whereas ionselective electrodes are used close to equilibrium conditions. This causes the oxygen electrode to be much more sensitive to changes in the temperature than potentiometric sensors. A typical application for this simple type of biosensor is the determination of glucose concentrations by the use of an immobilised glucose oxidase membrane. The reaction results in a reduction of the oxygen concentration as it diffuses through the biocatalytic membrane to the cathode, this being detected by a reduction in the current between the electrodes (Figure 19.3 ). Other oxidases may be used in a similar manner for the analysis of their substrates (e.g. alcohol oxidase, D- and L-amino acid oxidases, cholesterol oxidase, galactose oxidase, and urate oxidase) ________________________________________ Figure 19. 3. Schematic diagram of a simple amperometric biosensor. A potential is applied between the central platinum cathode and the annular silver anode. This generates a current (I) which is carried between the electrodes by means of a saturated solution of KCl. This electrode compartment is separated from the biocatalyst (here shown glucose oxidase, GOD) by a thin plastic membrane, permeable only to oxygen. The analyte solution is separated from the biocatalyst by another membrane, permeable to the substrate(s) and product(s). This biosensor is normally about 1 cm in diameter but has been scaled down to 0.25 mm diameter using a Pt wire cathode within a silver plated steel needle anode and utilising dip-coated membranes. ________________________________________ Figure 19. 4 An alternative method for determining the rate of this reaction is to measure the production of hydrogen peroxide directly by applying a potential of +0.68 V to the platinum electrode, relative to the Ag/AgCl electrode, and causing the reactions: Pt anode H2O2 O2 + 2H+ + 2e[19.6] Ag cathode 2AgCl + 2e- 2Ag0 + 2Cl-[19.7] The major problem with these biosensors is their dependence on the dissolved oxygen concentration. This may be overcome by the use of 'mediators' which
transfer the electrons directly to the electrode bypassing the reduction of the oxygen co-substrate. In order to be generally applicable these mediators must possess a number of useful properties. They must react rapidly with the reduced form of the enzyme. They must be sufficiently soluble, in both the oxidised and reduced forms, to be able to rapidly diffuse between the active site of the enzyme and the electrode surface. This solubility should, however, not be so great as to cause significant loss of the mediator from the biosensor's microenvironment to the bulk of the solution. However soluble, the mediator should generally be non-toxic. The overpotential for the regeneration of the oxidised mediator, at the electrode, should be low and independent of pH. The reduced form of the mediator should not readily react with oxygen. The ferrocenes represent a commonly used family of mediators (Figure 19.5 a). Their reactions may be represented as follows, Figure 19. 5 (a) Ferrocene (5-bis-cyclopentadienyl iron), the parent compound of a number of mediators. (b) TMP+, the cationic part of conducting organic crystals. (c) TCNQ.-, the anionic part of conducting organic crystals. It is a resonancestabilised radical formed by the one-electron oxidation of TCNQH2. Electrodes have now been developed which can remove the electrons directly from the reduced enzymes, without the necessity for such mediators. They utilise a coating of electrically conducting organic salts, such as N-methylphenazinium cation (NMP+, Figure6.7b) with tetracyanoquinodimethane radical anion (TCNQ.Figure 6.7c). Many flavo-enzymes are strongly adsorbed by such organic conductors due to the formation of salt links, utilising the alternate positive and negative charges, within their hydrophobic environment. Such enzyme electrodes can be prepared by simply dipping the electrode into a solution of the enzyme and they may remain stable for several months. Figure 19. 6 The response of an amperometric biosensor utilising glucose oxidase to the presence of glucose solutions. Between analyses the biosensor is placed in oxygenated buffer devoid of glucose. The steady rates of oxygen depletion may be used to generate standard response curves and determine unknown samples. The time required for an assay can be considerably reduced if only the initial transient (curved) part of the response need be used, via a suitable model and software. The wash-out time, which roughly equals the time the electrode spends in the sample solution, is also reduced significantly by this process. ________________________________________ These electrodes can also be used for reactions involving NAD(P)+-dependent dehydrogenases as they also allow the electrochemical oxidation of the reduced forms of these coenzymes. The three types of amperometric biosensor utilising product, mediator or organic conductors represent the three generations in biosensor development (Figure 19.6). The reduction in oxidation potential, found when mediators are used, greatly reduces the problem of interference by extraneous material. The current (i) produced by such amperometric biosensors is related to the rate of reaction (vA) by the expression: i = nFAvA (19.8) where n represents the number of electrons transferred, A is the electrode area, and F is the Faraday. Usually the rate of reaction is made diffusionally controlled by use of external membranes. Under these circumstances the electric current produced is proportional to the analyte concentration and independent both of the enzyme and electrochemical kinetics. Substrate(2H) + FAD-oxidase Product + FADH2-oxidasefi [19.9] This is followed by the processes: (a) biocatalyst FADH2-oxidase + O2 FAD-oxidase + H2O2 [19.10] electrode
H2O2 O2 + 2H+ + 2e[19.11] (b) biocatalyst FADH2-oxidase + 2 Ferricinium+ FAD-oxidase + 2 Ferrocene + 2H+ electrode 2 Ferrocene 2 Ferricinium+ + 2e[19.13] (c) biocatalyst/electrode FADH2-oxidase FAD-oxidase + 2H+ + 2e[19.14] ________________________________________
[19.12]
Figure 19. 7 Amperometric biosensors for flavo-oxidase enzymes illustrating the three generations in the development of a biosensor. The biocatalyst is shown schematically by the cross-hatching. (a) First generation electrode utilising the H2O2 produced by the reaction. (E0 = +0.68 V). (b) Second generation electrode utilising a mediator (ferrocene) to transfer the electrons, produced by the reaction, to the electrode. (E0 = +0.19 V). (c) Third generation electrode directly utilising the electrons produced by the reaction. (E0 = +0.10 V). All electrode potentials (E0) are relative to the Cl-/AgCl,Ag0 electrode. The following reaction occurs at the enzyme in all three biosensors: 19.5.3 Potentiometer biosensors Potentiometric biosensors make use of ion-selective electrodes in order to transduce the biological reaction into an electrical signal. In the simplest terms this consists of an immobilised enzyme membrane surrounding the probe from a pHmeter (Figure 19.8), where the catalysed reaction generates or absorbs hydrogen ions (Table 19.2). The reaction occurring next to the thin sensing glass membrane causes a change in pH which may be read directly from the pH-meter's display. Typical of the use of such electrodes is that the electrical potential is determined at very high impedance allowing effectively zero current flow and causing no interference with the reaction. ________________________________________ Figure 19. 8 A simple potentiometric biosensor. A semi-permeable membrane (a) surrounds the biocatalyst (b) entrapped next to the active glass membrane (c) of a pH probe (d). The electrical potential (e) is generated between the internal Ag/AgCl electrode (f) bathed in dilute HCl (g) and an external reference electrode (h). ________________________________________ A. There are three types of ion-selective electrodes which are of use in biosensors Glass electrodes for cations (e.g. normal pH electrodes) in which the sensing element is a very thin hydrated glass membrane which generates a transverse electrical potential due to the concentration-dependent competition between the cations for specific binding sites. The selectivity of this membrane is determined by the composition of the glass. The sensitivity to H+ is greater than that achievable for NH4+, Glass pH electrodes coated with a gas-permeable membrane selective for CO2, NH3 or H2S. The diffusion of the gas through this membrane causes a change in pH of a sensing solution between the membrane and the electrode which is then determined. Solid-state electrodes where the glass membrane is replaced by a thin membrane of a specific ion conductor made from a mixture of silver sulphide and a silver halide. The iodide electrode is useful for the determination of I- in the peroxidase reaction (Table 19.2c) and also responds to cyanide ions. ________________________________________ Table 19.2. Reactions involving the release or absorption of ions that may be utilised by potentiometric biosensors. (a) H+ cation,
glucose oxidase
H2O [19.5]
D-glucose + O2 D-glucono-1,5-lactone + H2O2 D-gluconate + H+ penicillinase penicillin penicilloic acid + H+ [19.6] urease (pH 6.0)a H2NCONH2 + H2O + 2H+ 2NH4+ + CO2 [19.7] urease (pH 9.5)b H2NCONH2 + 2H2O 2NH3 + HCO3- + H+ [19.8] lipase neutral lipids + H2O glycerol + fatty acids + H+ [19.9] (b) NH4+ cation, L-amino acid oxidase L-amino acid + O2 + H2O keto acid + NH4+ + H2O2 [19.20] asparaginase ` L-asparagine + H2O L-aspartate + NH4+ [19.21] urease (pH 7.5) H2NCONH2 + 2H2O + H+ 2NH4++ HCO3[19.22] (c) I- anion peroxidase H2O2 + 2H+ + 2I- I2 + 2H2O [19.23] (d) CN-anion -glucosidase amygdalin + 2H2O 2glucose + benzaldehyde + H+ + CN[19.24]
a Can also be used in NH4+ and CO2 (gas) potentiometric biosensors. b Can also be used in an NH3 (gas) potentiometric biosensor. ________________________________________ The response of an ion-selective electrode is given by (19.25) where E is the measured potential (in volts), E0 is a characteristic constant for the ion-selective/external electrode system, R is the gas constant, T is the absolute temperature (K), z is the signed ionic charge, F is the Faraday, and [i] is the concentration of the free uncomplexed ionic species (strictly, [i] should be the activity of the ion but at the concentrations normally encountered in biosensors, this is effectively equal to the concentration). This means, for example, that there is an increase in the electrical potential of 59 mv for every decade increase in the concentration of H+ at 25C. The logarithmic dependence of the potential on the ionic concentration is responsible both for the wide analytical range and the low accuracy and precision of these sensors. Their normal range of detection is 10-4 - 10-2 M, although a minority are ten-fold more sensitive. Typical response time are between one and five minutes allowing up to 30 analyses every hour. Biosensors which involve H+ release or utilisation necessitate the use of very weakly buffered solutions (i.e. < 5 mM) if a significant change in potential is to be determined. The relationship between pH change and substrate concentration is complex, including other such non-linear effects as pH-activity variation and protein buffering. However, conditions can often be found where there is a linear relationship between the apparent change in pH and the substrate concentration. A recent development from ion-selective electrodes is the production of ionselective field effect transistors (ISFETs) and their biosensor use as enzymelinked field effect transistors (ENFETs, Figure 19.9). Enzyme membranes are coated on the ion-selective gates of these electronic devices, the biosensor responding to the electrical potential change via the current output. Thus, these are potentiometric devices although they directly produce changes in the electric current. The main advantage of such devices is their extremely small size (<< 0.1 mm2) which allows cheap mass-produced fabrication using integrated circuit technology. As an example, a urea-sensitive FET (ENFET containing bound urease
with a reference electrode containing bound glycine) has been shown to show only a 15% variation in response to urea (0.05 - 10.0 mg ml-1) during its active lifetime of a month. Several analytes may be determined by miniaturised biosensors containing arrays of ISFETs and ENFETs. The sensitivity of FETs, however, may be affected by the composition, ionic strength and concentrations of the solutions analysed. ________________________________________ Figure 19. 9 Schematic diagram of the section across the width of an ENFET. The actual dimensions of the active area is about 500 m long by 50 m wide by 300 m thick. The main body of the biosensor is a p-type silicon chip with two n-type silicon areas; the negative source and the positive drain. The chip is insulated by a thin layer (0.1 m thick) of silica (SiO2) which forms the gate of the FET. Above this gate is an equally thin layer of H+-sensitive material (e.g. tantalum oxide), a protective ion selective membrane, the biocatalyst and the analyte solution, which is separated from sensitive parts of the FET by an inert encapsulating polyimide photopolymer. When a potential is applied between the electrodes, a current flows through the FET dependent upon the positive potential detected at the ion-selective gate and its consequent attraction of electrons into the depletion layer. This current (I) is compared with that from a similar, but non-catalytic ISFET immersed in the same solution. (Note that the electric current is, by convention, in the opposite direction to the flow of electrons). 19.5.4 Calorimetric biosensors Many enzyme catalysed reactions are exothermic, generating heat (Table 19.3) which may be used as a basis for measuring the rate of reaction and, hence, the analyte concentration. This represents the most generally applicable type of biosensor. The temperature changes are usually determined by means of thermistors at the entrance and exit of small packed bed columns containing immobilised enzymes within a constant temperature environment (Figure 19.10). Under such closely controlled conditions, up to 80% of the heat generated in the reaction may be registered as a temperature change in the sample stream. This may be simply calculated from the enthalpy change and the amount reacted. If a 1 mM reactant is completely converted to product in a reaction generating 100 kJ mole-1 then each ml of solution generates 0.1 J of heat. At 80% efficiency, this will cause a change in temperature of the solution amounting to approximately 0.02C. This is about the temperature change commonly encountered and necessitates a temperature resolution of 0.0001C for the biosensor to be generally useful. ________________________________________ Table 19.3. Heat output (molar enthalpies) of enzyme catalysed reactions. Reactant Enzyme Heat output -H (kJ mole-1) Cholesterol Cholesterol oxidase 53 Esters Chymotrypsin 4 - 16 Glucose Glucose oxidase 80 Hydrogen peroxide Catalase 100 Penicillin G Penicillinase 67 Peptides Trypsin 10 - 30 Starch Amylase 8 Sucrose Invertase 20 Urea Urease 61 Uric acid Uricase 49 ________________________________________ Figure 19. 10. Schematic diagram of a calorimetric biosensor. The sample stream (a) passes through the outer insulated box (b) to the heat exchanger (c) within an aluminium block (d). From there, it flows past the reference thermistor (e) and
into the packed bed bioreactor (f, 1ml volume), containing the biocatalyst, where the reaction occurs. The change in temperature is determined by the thermistor (g) and the solution passed to waste (h). External electronics (l) determines the difference in the resistance, and hence temperature, between the thermistors. ________________________________________ The thermistors, used to detect the temperature change, function by changing their electrical resistance with the temperature, obeying the relationship (19.26) therefore: (19.27) Where R1 and R2 are the resistances of the thermistors at absolute temperatures T1 and T2 respectively and B is a characteristic temperature constant for the thermistor. When the temperature change is very small, as in the present case, B(1/T1) - (1/T2) is very much smaller than one and this relationship may be substantially simplified using the approximation when x<<1 that ex1 + x (x here being B(1/T1) - (1/T2), (19.28) As T1 T2, they both may be replaced in the denominator by T1. (19.29) The relative decrease in the electrical resistance (R/R) of the thermistor is proportional to the increase in temperature (T). A typical proportionality constant (-B/T12) is -4%C-1. The resistance change is converted to a proportional voltage change, using a balanced Wheatstone bridge incorporating precision wirewound resistors, before amplification. The expectation that there will be a linear correlation between the response and the enzyme activity has been found to be borne out in practice. A major problem with this biosensor is the difficulty encountered in closely matching the characteristic temperature constants of the measurement and reference thermistors. An equal movement of only 1C in the background temperature of both thermistors commonly causes an apparent change in the relative resistances of the thermistors equivalent to 0.01C and equal to the full-scale change due to the reaction. It is clearly of great importance that such environmental temperature changes are avoided, which accounts for inclusion of the well-insulated aluminium block in the biosensor design . The sensitivity (10-4 M) and range (10-4 - 10-2 M) of thermistor biosensors are both quite low for the majority of applications although greater sensitivity is possible using the more exothermic reactions (e.g. catalase). The low sensitivity of the system can be increased substantially by increasing the heat output by the reaction. In the simplest case this can be achieved by linking together several reactions in a reaction pathway, all of which contribute to the heat output. Thus the sensitivity of the glucose analysis using glucose oxidase can be more than doubled by the co-immobilisation of catalase within the column reactor in order to disproportionate the hydrogen peroxide produced. An extreme case of this amplification is shown in the following recycle scheme for the detection of ADP. Figure 19. 11 ADP is the added analyte and excess glucose, phosphoenol pyruvate, NADH and oxygen are present to ensure maximum reaction. Four enzymes (hexokinase, pyruvate kinase, lactate dehydrogenase and lactate oxidase) are co-immobilised within the packed bed reactor. In spite of the positive enthalpy of the pyruvate kinase reaction, the overall process results in a 1000 fold increase in sensitivity, primarily due to the recycling between pyruvate and lactate. Reaction limitation due to low oxygen solubility may be overcome by replacing it with benzoquinone, which is reduced to hydroquinone by flavo-enzymes. Such reaction systems do, however, have the serious disadvantage in that they increase the probability of the occurrence of interference in the determination of the analyte of interest. Reactions involving the generation of hydrogen ions can be made more sensitive by the inclusion of a base having a high heat of protonation. For example, the heat output by the penicillinase reaction may be almost doubled by the use of Tris
(tris-(hydroxymethyl)aminomethane) as the buffer.In conclusion, the main advantages of the thermistor biosensor are its general applicability and the possibility for its use on turbid or strongly coloured solutions. The most important disadvantage is the difficulty in ensuring that the temperature of the sample stream remains constant ( 0.01C). 19.5.5 Immunosensors Biosensors may be used in conjunction with enzyme-linked immunosorbent assays (ELISA). The principles behind the ELISA technique is shown in Figure 16.9. ELISA is used to detect and amplify an antigen-antibody reaction; the amount of enzymelinked antigen bound to the immobilised antibody being determined by the relative concentration of the free and conjugated antigen and quantified by the rate of enzymic reaction. Enzymes with high turnover numbers are used in order to achieve rapid response. The sensitivity of such assays may be further enhanced by utilising enzyme-catalysed reactions which give intrinsically greater response; for instance, those giving rise to highly coloured, fluorescent or bioluminescent products. Assay kits using this technique are now available for a vast range of analyses. ________________________________________ Principles of a direct competitive ELISA. (i) Antibody, specific for the antigen of interest is immobilised on the surface of a tube. A mixture of a known amount of antigen-enzyme conjugate plus unknown concentration of sample antigen is placed in the tube and allowed to equilibrate. (ii) After a suitable period the antigen and antigen-enzyme conjugate will be distributed between the bound and free states dependent upon their relative concentrations. (iii) Unbound material is washed off and discarded. The amount of antigen-enzyme conjugate that is bound may be determined by the rate of the subsequent enzymic reaction. ________________________________________ Analyte Transducer/immobilization characteristics Low molecular weight analytes Surface plasmon resonance A monoclonal antibody against HBP is used. Lowest detection limit is 0.1 ng/ml, response time is 15 min DDT Surface plasmon resonance; covalent immobilization through an alkanethiol self-assembled monolayer (SAM) Lowest detection limit is 20 ng/L Organophosphorus Surface plasmon resonance; covalent immobilization through an alkanethiol self-assembled monolayer (SAM) Lowest detection limit is 50 ng/L Carbamate Surface plasmon resonance; covalent immobilization through an alkanethiol self-assembled monolayer (SAM) Lowest detection limit is 0.9 g/L CRP Surface plasmon resonance; Linear detection level is 2-5 g/ml using two different antiCPR antibodies Insulin with femtomole detection Liposomal immunosensors; fluorescence dye is used As low as 136 attomole. Total assay time is less than 30 min Antibiotics Poly and monoclonal antibodies are used. Recently ELISA techniques have been combined with biosensors, to form immunosensors, in order to increase their range, speed and sensitivity. A simple immunosensor configuration is shown in Figure 19.12 where the biosensor merely replaces the traditional colorimetric detection system. However more advanced immunosensors are being developed which rely on the direct detection of antigen bound to the antibody-coated surface of the biosensor. Piezoelectric and FET-based biosensors are particularly suited to such applications. ________________________________________ Figure 19. 12 Principles of immunosensors. (a)(i) A tube is coated with (immobilised) antigen. An excess of specific antibody-enzyme conjugate is placed in the tube and allowed to bind. (a)(ii) After a suitable period any unbound material is washed off. (a)(iii) The analyte antigen solution is passed into the tube, binding and releasing some of the antibody-enzyme conjugate dependent upon
the antigen's concentration. The amount of antibody-enzyme conjugate released is determined by the response from the biosensor. (b)(i) A transducer is coated with (immobilised) antibody, specific for the antigen of interest. The transducer is immersed in a solution containing a mixture of a known amount of antigen-enzyme conjugate plus unknown concentration of sample antigen. (b)(ii) After a suitable period the antigen and antigen-enzyme conjugate will be distributed between the bound and free states dependent upon their relative concentrations. (b)(iii) Unbound material is washed off and discarded. The amount of antigen-enzyme conjugate bound is determined directly from the transduced signal. Piezo-electric biosensors Piezo-electric crystals (e.g. quartz) vibrate under the influence of an electric field. The frequency of this oscillation (f) depends on their thickness and cut, each crystal having a characteristic resonant frequency. This resonant frequency changes as molecules adsorb or desorb from the surface of the crystal, obeying the relationshipes (19.30) where f is the change in resonant frequency (Hz), m is the change in mass of adsorbed material (g), K is a constant for the particular crystal dependent on such factors as its density and cut, and A is the adsorbing surface area (cm2). For any piezo-electric crystal, the change in frequency is proportional to the mass of absorbed material, up to about a 2% change. This frequency change is easily detected by relatively unsophisticated electronic circuits. A simple use of such a transducer is a formaldehyde biosensor, utilising a formaldehyde dehydrogenase coating immobilised to a quartz crystal and sensitive to gaseous formaldehyde. The major drawback of these devices is the interference from atmospheric humidity and the difficulty in using them for the determination of material in solution. They are, however, inexpensive, small and robust, and capable of giving a rapid response.
Chapter-19 Enzyme and protein STABILITY
Abstract:This report contains an overview of protein stability. The contribution each residue makes to, or takes away from, the stability of a protein is small. Thus the stability of a protein is determined by large number of small positive and negative interaction energies. In the sections that follow I discuss some of the factors that give rise to these positive and negative interaction energies. I will also briefly discuss rates of unfolding as they relate to perceived stability (Kinetic Stability),
I Definition of Stability Before entering into a discussion of stability in proteins, we must define exactly what we mean by stability. The word is used in different ways by different people. For example, a physical biochemist and a biotechnologist may each mean something different when they speak of stability. The physical biochemist, on the one hand, would probably discuss protein stability primarily in terms of the thermodynamic stability of a protein that unfolds and refolds rapidly, reversibly, cooperatively, and with a simple, two-state mechanism: Where Ku, is the equilibrium constant for unfolding. The easiest proteins in which to study folding and stability are those that exhibit this sort of rapid reversibility. Both experimental design and also theoretical treatment of data are simplified by reversible systems. Thus, it is no surprise that most of the literature reports about stability discuss this type of reversible system. The bulk of this dissertation will also focus on thermodynamic stability. In these cases, the stability of the protein is simply the difference in Gibbs free energy, G, between the folded and the unfolded states. The only factors affecting stability are the relative free energies of the folded (Gf) and the unfolded (Gu) states. The larger and more positive Gu, the more stable is the protein to denaturation. The Gibbs free energy, G, is made up the two terms enthalpy (H) and entropy (S), related by the equation: Where T is the temperature in Kelvin. The folding free energy difference, Gu, is typically small, of the order of 5- 15 kcal/mol for a globular protein (compared to e.g. ~30 - 100 kcal/mol for a covalent bond). The biotechnologist, on the other hand, is more concerned with the practical utility of the definition: Is the protein stable enough to function under harsh conditions of temperature or solvent? While the answer to this question may lie in thermodynamic stability (discussed above); it may also lie either simply in reversibility or, for irreversibly or slowly unfolding proteins, in kinetic stability. If a protein unfolds reversibly it may be fully unfolded and inactive at high temperatures, but once it cools to room temperature, it will refold and fully recover activity. From a functional standpoint this may be all that is required for it to be classified as thermostable. However, from a thermodynamic standpoint (and in terms of this dissertation) it is classified as non-thermostable. In the case of irreversible or slowly unfolding proteins, it is kinetic stability or the rate of unfolding that is important. A protein that is kinetically stable will unfold more slowly than a kinetically unstable protein. In a kinetically stable protein, a large free energy barrier to unfolding is required and the factors affecting stability are the relative free energies of the folded (Gf) and the transition state (Gts) for the first committed step on the unfolding pathway. Kinetic stability is discussed in more detail in its own section; see Kinetic Stability. Irreversible loss of protein folded structure is represented by: Where ki is the rate constant for some irreversible inactivation process. The free energy profile for a rapidly inactivating protein is shown below. Note that once the Unfolded form is reached, the energy barrier to inactivation is lower than that to refolding.
II The Unfolded State Until recently, most studies and theoretical treatments of protein stability have assumed that the unfolded state of a protein is a random coil, an ensemble of extended conformations, in which the protein chain is extensively hydrated and the individual residues do not interact with each other. This random coil ensemble can be treated as a single state for a thermodynamic description. One consequence of this view of the unfolded state is that any mutation that effects stability must have its effect almost exclusively via the native (folded) structure. However, there is now mounting evidence that this simplistic view of the unfolded state is not always true (reviewed by Shortle, 1996). For example, small angle Xray scattering studies have shown that the radius of gyration of the unfolded state of ribonuclease A is smaller than if it is in a random coil (Sosnick & Trewhella, 1992). Further, NMR studies have shown that interactions do occur between residues, particularly between aromatic residues, in the unfolded state and that these interactions can be non-native (e.g. Pan et al, 1995). In addition, there is strong evidence that mutations can affect the degree of interaction in the unfolded state (discussed in detail in Sturtevant, 1994). For example, in one mutant of Staphylococcal nuclease, the enthalpy of unfolding, H, was reduced to 50% that of wild-type, but with no concomitant change in G for the unfolding process (Tanaka et al, 1993). (Remember that ). Although possible, to explain this result in terms of changes in the folded state alone is difficult: requiring (i) that over half the stabilizing interactions present in the wild-type be absent in the mutant and (ii) that there be a huge increase in the entropy of the (folded) mutant over the native protein. A more convincing explanation of the result is that the enthalpy and entropy changes between mutant and wild-type are due to changes in the unfolded state, where introduction of a few interactions in the unfolded mutant could significantly decrease both enthalpy and entropy vis--vis the wild-type. There is further evidence of this type from unfolding studies with chaotropic denaturants which suggest that, for a small subset of mutants, the amount of change in accessible surface area upon unfolding is different to that in wild-type protein (Shortle & Meeker, 1986). To simplify assume that all of the intramolecular Van der Waals and hydrogen bonds that stabilize the native state are fully disrupted in the unfolded protein.
III Major Factors Affecting Protein Stability 1.hydrophobic interactions 2.hydrogen bonds 3.conformational entropy ________________________________________ The literature is in general agreement that the two types of interaction that are most prevalent in proteins are (i) hydrophobic interactions and (ii) hydrogen bonds. The reaction of these bonds upon going from the unfolded to the folded state is summarized in the cartoon below. Diagram showing the burial of hydrophobic moities and formation of intramolecular H-bonds upon protein folding. Note the release of water molecules upon folding. Although both these interactions have small free energies per residue, they are important because there are so many of them. The same is true for those interactions which stabilize the unfolded state. The most important of which is conformational entropy. Thus, the overall free energy of a folded protein is given by the small difference between two large numbers. This is a major reason for the difficulty of quantitative computational calculation of protein stability. In a
recent analysis of the factors contributing to the stability of RNase T1, the stabilizing and destabilizing interactions were estimated at 271 and 286 kcal/mol, respectively (Pace et al., 1996). Hydrogen bonding and hydrophobic interactions were estimated to contribute 260 kcal/mol to the stabilizing interactions, while the bulk of the destabilizing factors were attributed to loss of conformational entropy on folding, and unfavourable burial of peptide and polar groups. See table. Destabilising Free Energy (kcal/mol) Conformational Entropy -177 Peptide Groups Buried -81 Polar Groups Buried -28 Total Destabilising -286 Stabilising Histidine Ionisation +4 Disulphide Bonds +7 Hydrophobic Groups Buried +94 Hydrogen Bonding +166 Total Stabilising +271 ----------------------------------------------------------G (estimate) -15 G (measured) +9 Note that the while the difference between the experimental and estimated values is small, the estimated value would yield an unfolded protein. The Hydrophobic Effect The hydrophobic effect is considered to be the major driving force for the folding of globular proteins. It results in the burial of the hydrophobic residues in the core of the protein. It is exemplified by the fact that oil and water do not mix and was described well by G. S. Hartley in 1936 . "The antipathy of the paraffin chain for water is, however, frequently misunderstood. There is no question of actual repulsion between individual water molecules and paraffin chains, nor is there any very strong attraction of paraffin chains for one another. There is, however, a very strong attraction of water molecules for one another in comparison with which the paraffin-paraffin or paraffin-water attractions are slight." The thermodynamic factors which give rise to the hydrophobic effect are complex and still incompletely understood. The free energy of transfer of a non-polar compound from some reference state, such as an organic solution, into water, Gtr, is made up of an enthalpy, H, and entropy, -T S, term. At room temperature, the enthalpy of transfer from organic solution into aqueous solution is negligible; the interaction enthalpies are the same in both cases. The entropy however is negative. Water tends to form ordered cages around the nonpolar molecule and this leads to a decrease in entropy. At high temperatures (~ 110C) these cages are no longer any stronger than bulk water, and the entropy contribution tends to zero. The enthalpy of transfer, however, is now positive (unfavourable). Because the temperature dependence of entropy and enthalpy are not the same, there is some temperature at which the hydrophobic effect is strongest, and the effect decreases at temperatures above and below this temperature. The decrease in the strength of the hydrophobic effect with decreasing temperatures is probably the major cause of cold-denaturation in proteins. The contribution of the hydrophobic effect to globular protein stability has been estimated empirically both by measuring the thermodynamics of transfer of model compounds (e.g. blocked amino acids, cyclic peptides...) from organic solvents to water, and by site directed mutagenesis studies on proteins. The number arrived at
is usually given as a function of the change in the solvent accessible non-polar surface area upon going from the unfolded to the folded state. The model compound studies predict that the hydrophobic effect of exposing one buried methylene group to bulk water is 0.8 kcal/mol (in Pace, 1995). The site directed mutagenesis studies yielded a larger number with greater statistical variation: the average hydrophobic effect estimated by SDM for a buried methylene group is about 1.3 kcal/mol. However, when the SDM results for methylene were plotted against the size of the cavity created by the residue substitution, and extrapolated to zero, the result at zero cavity size is 0.8 kcal/mol - in agreement with the value found for the transfer of model compounds from octanol to water (Pace et al., 1996 and references therein). In the SDM studies, cavities created by residue substitution have an additional destabilizing effect: the loss of favourable VDWs interactions (as compared to the wild-type). Thus, the "hydrophobic effect" measured by SDM includes both an entropic component due to solvent ordering and a (primarily) enthalpic component due to loss of VDWs contacts within the protein. Such an SDM study of T4 lysozyme replaced the 80% buried Ile3 residue by Val (Eriksson et al, 1992): the loss of this methyl group gave rise to a decrease in stability of 0.6 kcal/mol (corrected to 100% burial). This is smaller than expected (c.f. 0.8 kcal/mol for methylene) and suggests that the mutation introduced some smaller stabilizing influence, perhaps such as the alleviation of strain within the protein. In barnase, 15 mutants were constructed in which a hydrophobic interaction was deleted (V10A, V36A, V45A, I4A, I25A, I51A, I55A, I76A, I109A, I4V, I25V, I51V, I55V, I76V & I109V). The finding was a strong correlation between the degree of destabilization (which ranges from 0.60 to 4.71 kcal/mol) and the number of methyl or methylene side chain groups surrounding the methyl or methylene group that was deleted (r = 0.91) (Serrano et al, 1992). See figure below. Correlation between the number of side chain methylene and methyl groups, in a radius of 6 of the group deleted from wild-type, and the changes in the free energy of unfolding for mutations of hydrophobic residues in barnase. (Taken from Serrano et al, 1992. with permission) The average free energy decrease for removal of a completely buried methylene group was found to be 1.50.6 kcal/mol. This is additive, such that Ile or Leu to Ala can destabilize a protein by up to 5 kcal/mol. (Remember that many mesophilic proteins are stable by <10 kcal/mol, so two deletions such as this would be enough to destabilize a protein completely). Hydrogen Bonds A hydrogen bond occurs when two electronegative atoms, such as nitrogen and oxygen, interact with the same hydrogen. The hydrogen is normally covalently attached to one atom, the donor, but interacts electrostatically with the other, the acceptor. This interaction is due to the dipole between the electronegative atoms and the proton. There is a geometric component involved in hydrogen bonds, and for single donor acceptor systems, such as N-H---O, the strongest hydrogen bonds are collinear (Creighton, 1993 and references therein). Electrostatic calculations suggest that deviation of 20 from linearity leads to a decrease in binding energy of approximately 10% (Pimentel & McClellan, 1960). In double acceptor systems, bifurcated hydrogen bonds with non-linear angles are preferred. The occurrence of hydrogen bonds in protein structure has been extensively reviewed by Baker & Hubbard (1984), albeit before the pdb database was as large as it is today. They found that 90% of N-H---O bonds in proteins lie between 140 and 180, and that they are centred around 158C. For C=O---H, the range is more broadly distributed between 90 and 160 and centred around 129. The strength of a hydrogen bond is between 2 and 10 kcal/mol, and one might think
that this is the amount of energy one hydrogen bond contributes towards stabilization of a folded protein. However, in the unfolded state, all potential hydrogen bonding partners in the extended polypeptide chain are satisfied by hydrogen bonds to water. When the protein folds, these protein-to-water H-bonds are broken, and only some are replaced by (often sub-optimal) intra-protein Hbonds. McDonald & Thornton (1994) showed that while only 1.3% of backbone amino groups and 1.8% of carbonyl groups in proteins fail to H-bond (without any obviously compensating interactions), 80% of main chain carbonyls fail to form a second hydrogen bond. Thus, if one considers enthalpy terms alone, it would appear that hydrogen bonding is destabilizing to folded protein structure. However, one must also consider entropy. When a protein folds, and those hydrogen bonds that the protein made to bulk water are broken, the entropy of the solvent increases. The balance between the entropy and enthalpy terms are close, and in the recent past it was considered that H-bonds made no contribution overall to protein stability. But, it is now generally accepted that H-bonds make a positive contribution to protein stabilisation (reviewed in Pace et al., 1996). Estimation of the contribution of hydrogen bonding to protein stability has been made by a combination of experiments on model compounds and site-directed mutational (SDM) studies. The difficulty with the SDM studies is that when a smaller residue replaces a larger one, a cavity is created. For example, mutating Asn to Ala creates a cavity of 37.4 3 (Harpaz et al , 1994). This cavity may then be filled by water, replacing the hydrogen bonding of the asparagine NH group. Even in more conservative mutations such as Thr to Val (which is isosteric) or Ser to Ala, one must take into account the contribution of side chain entropy and the hydrophobic effect to the ( G) values. Dissection of the latter contributions from those due only to hydrogen bonds is not trivial. An estimation has been made of a positive contribution of 1.51.0 kcal/mol (Pace et al., 1996, Fersht, 1987) from the formation of a buried intramolecular uncharged hydrogen bond. However, in order to form, the unfavourable interaction energy from burial of a polar group must be overcome. Thus, the net energy gain for formation of a buried H-bond is approximately 0.6 kcal/mol. Despite the small contribution made to protein stability by hydrogen bonds, we must remember that if we break or delete an intramolecular hydrogen bond in a protein without the possibility of forming a compensating H-bond to solvent, that protein will be destabilized. In globular proteins, much of the H-bonding potential of the backbone amide and carbonyl groups is satisfied by the formation of regular structure such as alpha helix and beta sheet (links to PPS); regular structure comprises 80 - 90% of globular protein structure. There is evidence that hydrogen bonds contribute to stability in hyperthermostable proteins. A comparison of glyceraldehyde-3-phosphate dehydrogenase (GADH) from four organisms with a range of thermostabilities and more than 50% sequence identity found that the strongest correlation to thermostability was with the number of buried charged residues H-bonded to buried neutral residues (Tanner et al, 1996). The rationalization given for this preference of charged-to-neutral over neutral-to-neutral or charged-to-charged residue H-bonding was as follows: The enthalpy of H-bond formation is in the order, charged-to-charged > charged-toneutral > neutral-to-neutral, but the entropic cost of desolvation is in the inverse order. The greatest overall free energy benefit is proposed to be for the charged-to-neutral H-bonds. Conformational Entropy of Unfolding The factor that makes the greatest contribution to stabilization of the unfolded state is its conformational entropy. It has been proposed that decreasing the conformational flexibility of the unfolded chain (by substitution with proline, or by replacement of glycine) should lead to an increase in the stability of the folded relative to the unfolded protein (Matthews et al, 1987) .Two such substitutions (A82P and G77A) in T4 lysozyme gave rise to only a small amount of stabilization (0.8 and 0.4 kcal/mol), possibly due to the counterbalancing removal
of favourable interactions upon mutation.(Matthews et al, 1987) A similar mutation that also eliminated a hydrogen bond and some hydrophobic interactions was destabilizing, suggesting that these factors outweighed the decrease in conformational stability (Dixon et al, 1992). Watanabe et al (1994) have found a correlation between the number of proline residues in oligo-1,6-glucosidases from a number of bacterial species and their thermostability. Structural analysis suggests that the optimal placement is at the N-cap of alpha helices and at the second position beta type I and beta type II turns. When they substituted prolines at these positions in the homologous mesophilic enzymes they observed an increase in thermostability. However, they made their substitutions based on recruitment from the more stable enzyme. When I tried this approach in the thermostable alpha amylase from Bacillus lichinoformis, based on structural parameters alone, the mutants were either equal in stability to wildtype or destabilized (A. Day, unpublished). Structural analysis of at least one destabilized mutant (A. Day & A. Shaw unpublished) revealed the creation of a hydrophobic surface cavity upon mutation, which presumably counterbalanced any stabilizing effect of the conformational rigidity. This suggests that, in the absence of any hints from nature in terms of homology, proline substitution for stabilization should be used with some care. Watanabe et al ascribe their increase in stability to a decrease in conformational entropy of the unfolded state (c.f. the T4 lysozyme case described above by Matthews et al, 1987). As their enzyme (and ours) is irreversibly inactivated upon heating, it is equally likely that any observed increase in stability is due, not to an overall stabilization of the protein, but to a slowing of the rate of unfolding. IV Other Factors Affecting Protein Stability There are a number of other interactions, in addition to those already discussed (The Hydrophobic Effect, Hydrogen Bonds, Conformational Entropy of Unfolding), which make contributions to the stability of proteins. These interactions are few relative to the major factors, but are still have some importance for protein stability. They include: Salt Bridges Aromatic-Aromatic Interactions Metal Binding and Disulphide Bonds Salt Bridges Salt bridges or ion-pairs are a special form of particularly strong hydrogen bonds made up of the interaction between two charged residues. The contribution of salt bridges to protein stability is a somewhat contentious issue in the literature. On the one hand is the observation that thermophilic and hyper-thermophilic analogues of mesophilic proteins tend to have increased numbers of salt-bridges (Tanner et al., 1996; Perutz & Raidt, 1975; Perutz, 1978; Dekker et al., 1991). On the other hand are mutational studies showing that the contribution of salt bridges to stability is small. Perhaps at higher temperatures salt bridges make more of a contribution to stability. Horovitz et al . (1990) measured the stability of a surface salt bridge triad between Asp8, Asp12 and Arg110 on the surface of barnase by construction of a thermodynamic cycle of all possible combinations of 1, 2, or 3 alanine mutants. The free energy contribution to the stability of the protein is only 1.25 kcal/mol for the Asp12/Arg110 pair and 0.98 kcal/mol for the Asp8/Arg110 pair. Removal of Arg 110 has no effect on stability! Other studies have had similar results (Akke & Forsen, 1990). One reason that these contributions are not as large as might be expected from the strength of such an ion pair is that, in order form a salt bridge, strong hydrogen bonds with
water have to be broken. In fact, it is possible that most of the energy of stabilization comes from the increase in solvent entropy upon formation of the ion pair. In contrast to surface salt bridges, removal of one partner from a buried salt bridge leads to destabilization of 3-4 kcal/mol. However, it has been found that replacing a buried salt-bridge triad by well packed hydrophobic residues (found by random mutagenesis at the three charged residue positions) leads to an increase in stability of 4.5 kcal/mol in the arc repressor (Waldburger et al., 1995). Thus, hydrophobic interactions contribute more to stability than a salt bridge triad. This is illustrated below and is presumably due to the cost of desolvating the charged groups on going from the unfolded to the folded state. The mutant is R31M, E36Y, R40L. Interestingly, it was also shown that the wild-type arc repressor folds between 10 and 1250 more slowly than the mutant (Waldburger et al., 1996). This is proposed to be due to the high energy barrier to burying charged residues. The transition state would have a particularly high energy if one charged residue had to be buried before the other. Even if the salt bridge had formed prior to the transition state, the geometry of the salt bridge might well be sub-optimal until that part of the protein attained its native conformation. Hydrophobic interactions have less of a steric requirement. Aromatic-Aromatic Interactions About 60% of the aromatic side chains (Phe, Tyr, and Trp), found in proteins are involved in aromatic pairings. Studies with model compounds suggest that the optimal geometry is perpendicular, such that the partially positively charged hydrogens on the edge of one ring can interact favourably with the pi electrons and partially negatively charged carbons of the other. From these observations, it might be expected that such interactions make a contribution to protein stability. This has been tested on the solvent exposed Tyr13 / Tyr17 pair on the surface of barnase (Serrano et al., 1991). Tyr13 and Tyr17 were mutated to both Ala and Phe. Mutation of the Tyr13 or Tyr17 to Phe leads to a decrease in stability of 0.30 or 0.41 kcal/mol, and 0.61 kcal/mol in the double mutant. As both hydroxyl groups are exposed to solvent (as they would be in the unfolded state), it is unclear as to the source of this small destabilization. However, mutation to the double alanine mutant leads to a decrease in stability of 4.6 kcal/mol. Most of this destabilization can be explained in terms of the loss of interactions between the tyrosine residues and the rest of the protein; however, analysis of the data using thermodynamic cycles (Carter, 1984; Horovitz & Fersht, 1990) indicates that the interaction energy between the two aromatic groups contributes only 1.3 kcal/mol to the protein stability. This is only very slightly higher than the stabilization expected from the hydrophobic contribution from burying surface area between them. In this case, therefore, there is little apparent extra stabilization due to the aromatic pair. Metal Binding Another method by which the folded state of proteins can be stabilized is metal binding, in which metal ions are coordinated, usually by lone pair donation from oxygen or nitrogen atoms. Experiments have shown that metal binding can contribute 6 - 9 kcal/mol (Braxton, 1996 and references therein) to stability. However, this is a little misleading as the comparisons made are between the apo- and the holo- enzyme; in the apo-enzyme there is often a destabilising cluster of negative charge from coordinating acidic side chains. Perhaps a fairer estimation of the contribution of metal binding to protein stability comes from an experiment in which a metal chelating site was introduced into an alpha helix of iso-cytochrome c and gave rise to a 1 kcal/mol increase in stability in the presence of saturating Cu(II) (Kellis et al., 1991). Another interesting study was that of Kuroki et al. (1989). They observed that the sequence and tertiary structure of alpha-lactalbumin (Ca2+ binding) and c-type
lysozymes (non-Ca2+ binding) are homologous. They recruited the binding site from alpha-lactalbumin into human c-type lysozyme (Q86D/A92D). The mutant protein binds one mole of calcium ions and has optimal activity at about 10C higher than the wild-type. Interestingly, the apo-enzyme is about 5 C less stable than wild-type. Disulphide Bonds Disulphide bonds are formed by the oxidation of two cysteine residues to form a covalent sulphur-sulphur bond which can be intra- (examples are shown in Jane Richardson's Protein Tourist kinamage) or inter- (exemplified by insulin at this PPS link) molecular bridges. One might imagine that as the enthalpy of a covalent disulphide bond is very high, it contributes a great deal to stability. However, this bond is present in both the folded and the unfolded state, thus its enthalpic contribution to the free energy difference is negligible. All of the stabilizing effect of a disulphide bond is proposed to come from the decrease in conformational entropy of the unfolded state, as described in Conformational Entropy of Unfolding, above. Calculations suggest that a disulphide bond should give rise to 2.5 - 3.5 kcal/mol of stabilization, depending on the primary sequence separation between the crosslinks (Braxton, 1996). Experiments in which naturally occurring disulphides are either mutated to alanine, or chemically reduced and blocked, lead to decreased stability ranging from 2 - 8 kcal/mol (Betz, 1993 and references therein). Unfortunately, because these experiments also leave cavities, or buried polar groups, or otherwise have more consequences than just removal of the disulphide bridge, it is hard to estimate the stabilization due to crosslinks alone. Introduction of novel disulphides into proteins has also had mixed results. Five disulphides have been introduced into T4 lysozyme (which has no disulphides in the wild-type). Two are destabilizing, and three are stabilizing relative to their reduced form, but all five are destabilized relative to wild-type (Betz, 1993)! However, there have been successful stabilizations by introduction of disulphides: The stability of RNase Hn is increased by 2.8 kcal/mol upon introduction of a disulphide. Engineering disulphides into the ribonuclease barnase gave rise to increased stability of 1.2 and 4.1 kcal/mol (Clarke et al., 1995). Interestingly, the former disulphide encompasses a loop of 34 residues, while the latter encompasses 17. If the stabilization is purely due to conformational entropy, the magnitude of stabilization would be the opposite way round; further, the degree of stabilization observed for the 17 residue loop (4.1 kcal/mol) is higher than would be predicted by the theory (Betz, 1993 ). Thus, it is clear that we are still far from understanding the role of disulphides in protein structure. It is worth noting that small proteins are often naturally rich in disulphide bonds, perhaps, when the geometry is optimal, they compensate for the small number of non-covalent interactions. Disulphide linkages are rare in hyper-thermostable proteins, presumably because they are chemically labile at high temperatures . Disulphides can also contribute a great deal to Kinetic Stability . V Chemical Degradation It should be mentioned that however stable a protein can be made by stabilizing the folded state, the ultimate limit of protein stability must come from covalent degradation. At high temperatures (80 - 120 C) Asn and Gln are susceptible to deamidation, Asp-Xaa peptide bonds are susceptible to hydrolysis, disulphides bonds rupture, and Xaa-Pro peptide bonds undergo cis-trans isomerisation (where Xaa is any amino acid). Interestingly, the upper limit for protein chemical thermostability may be higher than one would calculate from studies involving model mesophilic enzymes. Apart
from the trivial response of just avoiding these residues (disulphides are absent and Asn/Gln content is reduced in hyper-thermophiles), it is observed that the deamidation rate of Gln and Asn residues is reduced, presumably by steric constraint, in fully folded hyper-thermophilic structures (Vielle & Zeikus, 1996 and references therein). VI Conclusions This dissertation has discussed some of the many forces that make small and conflicting contributions to protein stability. It is the sum of these various stabilising and destabilising interactions that gives rise to the final stability of a protein. The total destabilising and the total stabilising energies are both large, and their difference is small. This is one of the reasons that current computational methods struggle to predict protein stability from structure. Furthermore, our understanding of these many forces is incomplete; as often as not mutations have the opposite effect to that which had been predicted. In addition, the activation energy for folding is an important determinant of both kinetic stability and whether a protein will fold to a global minimum. However, it also clear that great strides have been made, and the search for the solution to The Folding Problem, one of the great remaining questions in biology, is and continues to be, an exciting one.
Chapter -20 Study of drug stratgies
INTRODUCTION DNA is the carrier of all genetic information in most organisms and thus a biological molecule of paramount importance. DNA replication and transcription are the basic steps in the processes of cell division and gene expression. DNA replication and transcription are regulated by several small DNA binding proteins such as transcription factors and polymerases. A number of molecules have been artificially created that mimic the interactions between the DNA and these regulatory proteins. These molecules have served as useful drugs that can be used to inibit, activate or modulate the processes of DNA replication and transcription by binding to the DNA instead of the regulatory proteins. Though the actions of activation and modulation are possible, mostly DNA is targeted in an
inhibitory mode, to destroy cells for antitumor and antibiotic action. TYPES OF DNA BINDING DRUGS We are already aware of the Watson and there are two sugar phosphate backbone in an antiparallel fashion and between other. DNA binding drugs interact with NON COVALENT INTERACTIONS Crick model of DNA double helix in which strands running spirally around each other them base pairs stacked one above the DNA either non-covalently or covalently.
Non covalent interactions are generally reversible. Drugs that undergo non covalent interactions with the DNA can be classified into two main classes: Minor Groove binders Intercalators MINOR GROOVE BINDERS Minor groove binding drugs are usually shaped such that they easily fit into the groove. The binding mainly is promoted by van der Waals interactions. These drugs can also form hydrogen bonds with N-3 of adenine and O-2 of thymine. Generally minor groove binding has negligible influence on the conformation of DNA. Most minor groove binding drugs bind to A/T rich sequences. Example: Berenil. Recently, a few synthetic polyamides like lexitropsins and imidazole-pyrrole polyamides specific forG-C and C-G regions in the grooves have been designed. These polyamides are linked systems that recognize DNA base pairs through specific orientation of imidazole (Im) and pyrrole (Py) rings. Example: The eight ring hairpin polyamide ImPyPyPy-g-ImPyPyPy-b-Dp (Dp dimethylamino propylamide) has been found to inhibit the expression of 5S RNA in fibroblast cells (skin cancer cells) by blocking the transcription factor IIIAbinding site. INTERCALATORS Intercalating agents contain planar heterocyclic groups which plug in between adjacent DNA base pairs. The complex, thus formed, is mainly stabilized by - stacking interactions between the drug and DNA bases. Triostin A has two quinoxaline units connected by a cyclic peptide structure with a cystein pair (disulfide bond) at its center. Chemical structure of triostatin A
Triostin A is an antibiotic possesing cross-linked octapeptide rings attached to two quinoxaline chromophores. The chromophores spaced at 3.5A, sandwich two base pairs between the two quinoxalines. Intercalators induce strong structural distortions in DNA. For example, Daunomycin is a DNA intercalator that is site specific for GC base pairs. On intercalation of DNA by Daunomycin the following structural changes occur: CG and GC base pairs bend by ca. 9o and 15o respectively to prevent excessive van der Waals contacts.
Also, the base pairs are wedged out to a separation of 6.8 . These distortions lead to a total DNA unwinding angle of ca. 8o. These changes have the effect of inhibiting the association of DNA with the DNA helicase and topoisomerase.
COVALENT INTERACTIONS Covalent binders bind the DNA irreversibly, invariably leading to complete inhibition of DNA processes and subsequent cell death Important drugs belonging to this category are: Mitomycin C - antitumor antibiotic The activated antibiotic forms a cross links guanine bases on adjacent strands of DNA thereby inhibiting single strand formation. Anthramycin - antitumor antibiotic It binds covalently to N-2 of guanine located in the minor groove of DNA. Cisplatin (cis-diamine-dichloro-platinum) - anticancer drug. It platinates N-7 of guanine on the major groove site of DNA double helix. This leads to the cross-linking of two adjacent guanines on the same DNA strand hindering the mobility of DNA polymerases. FACTORS AFFECTING DRUG-DNA INTERACTIONS
BASE PAIR SEQUENCE Base pair sequence plays a large role on the specific nature of most DNA binding drugs. The nature and strength of the interaction occurring between the drug and the DNA depends greatly upon the the specific bases involved. The predisposition of minor groove binders like Berenil towards A/T rich regions could be explained by the following facts: A/T regions are narrower than G/C regions, thus, better van der Waals contacts. High steric hindrance in the G/C groove regions, due to the C-2 amino group of the guanine base. In the case of intercalator triostin A it has been proposed that the linker peptide structure could be responsible for specific interaction with the DNA surface. The major group specific readout sequence of H-bond donor and acceptor could be involved in triostatin A binding. COUNTER IONS DNA carries a large negative charge because of the phosphate backbone. Therefore, small positively charged counter ions such as Na+, or Ca++ and Mg++ ions may be present around the DNA. The counter ions can effectively screen the negative backbone surface thereby allowing non electrolytes as well as positively charged drug ligands to interact more strongly with the target base pairs. SOLVENT In the process of drug DNA binding, a displacement of solvent from the binding site on both the DNA and drug takes place. There also occurs a partial compensation of charges as the DNA and drug are oppositely charged which causes some partially solvated counterions to be released into the bulk solvent and become fully solvated.
Hydrophobic effect is the major driving force of hydrophobic ligand receptor interaction. It is seen in the case of intercalating drugs as the hydrophobic, aromatic sidechains interact favorably with the aromatic environment of the base pair stacking. STRUCTURAL MODIFICATIONS So that the DNA and the drug molecule can accommodate each other, some structural deformation/adaptation occurs in both. Thus, it can be concluded that some of the important forces contributing to drug DNA interactions are- hydrophobic force, hydration/dehydration energies, van der waals interactions and hydrogen bonds, ion effects, etc.
CHEMOTHERAPY- DNA BINDING DRUGS AGAINST CANCER Cancer cells exhibit rapid cell division. They lose the property of contact inhibition and do not undergo appoptosis. Chemotherapy makes use of DNA binding drugs to kill cancerous cells. The chemotherepeutic drugs bind to the DNA of the cancer cells and halt the cell division. When the cancer cells fail to divide they die, ultimately causing the entire tumor to shrink. Some of the important DNA binding drugs used in chemotherapy are cisplatin, amifosatine and mitomycin C. SIDE EFFECTS OF CHEMOTHERAPY Current mechanisms target chemotherapeutic drugs to cells that are rapidly divivding. Unfortunately, chemotherapy cannot differentiate cancer cells from healthy cells. As a result, fast dividing healthy cells such as stem cells also become severely affected by the drugs. Side effects caused by chemotherapt may be from mild to severe such as: Hair loss Nausea Low RBC count Decreased immunity Faliure of certain organs Even death. COUNTERACTING SIDE EFFECTS DESIGNING MORE ACCURATE DRUG DELIVERY STRATEGIES* For designing better drug delivery strategies, we should focus upon the differences between cancer cells and normal cells at olecular level. Conventional methods target rapidly dividing cells. The disadvantge of this approach is that, it cannot distinguish cancer cells from the stem cells, which leads to a number serious side effects. The properties of cancer cells that are unique to them should be exploited for drug targetting. A few moleculare features of cancer cells that may serve as molecular targets in chemotherapy are listed belowCancer cells have been found to be much less adhesive to other cells and non cellular substrates. It is this lack of adhesiveness that permits cancer cells to
metastasize. This happens because of the expression of new cell surface proteins reffered to as tumor associated antigens in cancer cells that can induce modiications at the cell surface, thereby leading to the loss of adhesiveness. Cancer cells continue to divide indefinitely, without undergoing ageing this seemingly immortal nature of cancer cells can be attributed to the presence of the telomerase enzyme which is absent in normal cells. The telomerase enzyme matains teloeres at the ends of the chromosomes, thus allowing cells to continue to divide. Cancer cells require large amounts of glucose for energy production and growth. When cells become cancerous, they require more energy and the level of proteins needed for glucose transport and metabolism increases. As a tumor grows, it rapidly outgrows its blood supply, leaving portions of the tumor with regions where the oxygen concentration is significantly lower than in healthy tissues. As a consequence, tumor cells in these hypoxic zones rely on glycolysis for energy production and therefore further increase the levels of proteins responsible for glucose transport and metabolism. These differences could be employed for transpoting the DNA binding drugs to the cancer cells without affecting the healthy cells of the body. DESTABLISING DRUG DNA INTERACTIONS IN HEALTHY STEM CELLS* A DNA binding drug binds with the DNA based on simple rules of chemistry. The interactions that stablise most drug-DNA complexes are simple hydogen bonds, hydrophobic effects and electrostatic interations. The whole concept rounds upto the fact that the product (drug DNA complex) must be more stable than the starting elemnt - unbound drug. If we could design synthetic molecules that offered a particular drug a set of more stablising interactions inside the cell then it could be thought of using this molecule to destablise the drug-DNA interactions in the healthy cells so that they can resume their normal cell cycle. Let us consider a situation for understanding this conept better. Let us assume that a patient of cancer was administerred with anthracycline(an inercalator), which due to inefficient drug delivery found its way into stem cells and got bound to their DNA. Now, side effects begin to manifest. In order to nullify the side effects a synthetic molecule is designed that provides a greater affinity to the drug bound to stem cell DNA than the DNA itself. This synthetic agent can now be targetted to the stem cells where it will act so as to pull anthracycline from the DNA and itself form a stabler complex with it. Now the stem cells are free of the drug and can undergo their normal cellular functions.
CONCLUSION Thus, we see that DNA binding drugs of all categories have displayed a promising potential in the treatment various deadly viral diseases and cancers. For this reason they have become targets of clinical research across the globe. DNA binding drugs such as cis-Platin have revolutionised chemotherapy so that now it is possible to completely root out cancers. However, there several hurdles
still to be cleared before these drugs can be considered as a safe cure for cancer. The scientific community understands this and therefore full fledged research work is being conducted in several big research institutes and laborataries to design better and safer drugs.
SUMMARY
Certain molecules are able to mimic the naturally occuring DNA binding proteins such as transcription factors and DNA polymerases. These molecules are used as drugs that can be bound physically to the DNA for interrupting key cellular activities such as DNA replication and gene expression. DNA drug interactions can be covalent or non covalent. DNA binding drugs can be grouped into 3 categories minor groove binders, intercalators and covalent binders. Important forces contributing to drug DNA interactions are- hydrophobic force, hydration/dehydration energies, van der waals interactions and hydrogen bonds, ion effects, etc Sveral DNA binding drugs have played an invaluable role in cancer chemotherapy, such as cis-Platin, daunomycin, and so on. Side effects of chemotherapy can be minimised by designing strategies for efficient drug targetting and destabilising the drug-DNA complexes in healthy cells. Assignment Structural Biology DNA And Drug Interaction
Introduction to DNA Deoxyribonucleic acid, DNA, is a molecule of great biological significance. The total DNA content of a cell is termed the Genome. The Genome is unique to an organism, and is the information bank governing all life processes of the organism, DNA being the form in which this information is stored. Stretches of DNA called genes have the extremely important function of coding for proteins. The function of the rest of the genome, loosely termed as non-gene regions, is not very clearly known. Fig.1 The DNA molecule
DNA has two main functions, 1. Transcription: Information is retrieved from the DNA by ribonucleic acid, RNA, and utilized to synthesize proteins in the body. Proteins are involved in all body processes and play many roles. e.g. as hormones, enzymes, carriers, structural proteins, receptors, regulators etc.
2. Replication: DNA is responsible for its own regeneration, i.e., DNA self replicates. DNA is present in the body in the form of a double helix, where each strand is composed of a combination of four nucleotides, adenine (A), thymine (T), guanine (G) and cytosine (C). Within a strand these nucleotides are connected via phosphodiester linkages. The two strands are held together primarily via Watson Crick hydrogen bonds where A forms two hydrogen bonds with T and C forms three hydrogen bonds with G (Figure2). fig.2 Watson Crick Base pairing, A-T and G-C base pairing Specific recognition of DNA sequences by proteins/ small molecules is achieved via the combination of hydrogen bond acceptor/donor sites available on the major groove or minor groove. e.g. the A-T base pair offers a hydrogen bond acceptor, N7, a donor N6, and an acceptor, O4 on the major groove side. Transcription and replication are vital to cell survival and proliferation as well as for smooth functioning of all body processes. DNA starts transcribing or replicating only when it receives a signal, which is often in the form of a regulatory protein binding to a particular region of the DNA. Thus, if the binding specificity and strength of this regulatory protein can be mimicked by a small molecule, then DNA function can be artificially modulated, inhibited or activated by binding this molecule instead of the protein. Thus, this synthetic/natural small molecule can act as a drug when activation or inhibition of DNA function is required to cure or control a disease (Table 1). DNA and Drug Binding DNA activation would produce more quantities of the required protein, or could induce DNA replication; depending on which site the drug is targeted. DNA inhibition would restrict protein synthesis, or replication, and could induce cell death. Though both these actions are possible, mostly DNA is targeted in an inhibitory mode, to destroy cells for antitumor and antibiotic action. Drugs bind to DNA both covalently as well as non-covalently. Covalent Binding Drugs Covalent binding in DNA is irreversible and invariably leads to complete inhibition of DNA processes and subsequent cell death. Cis-platin (cisdiamminedichloroplatinum) is a famous covalent binder used as an anticancer drug, and makes an intra/interstrand cross-link via the chloro groups with the nitrogens on the DNA bases. Cisplatin, cisplatinum or cis-diamminedichloroplatinum(II) (CDDP) is a platinumbased chemotherapy drug used to treat various types of cancers, including sarcomas, some carcinomas (e.g. small cell lung cancer, and ovarian cancer), lymphomas and germ cell tumors. It was the first member of its class, which now also includes carboplatin and oxaliplatin.
Upon administration, a chloride ligand undergoes slow displacement with water (an
aqua ligand) molecules, in a process termed aquation. The aqua ligand in the resulting [PtCl(H2O)(NH3)2]+ is easily displaced, allowing cisplatin to coordinate a basic site in DNA. Subsequently, the platinum cross-links two bases via displacement of the other chloride ligand.[1] Cisplatin crosslinks DNA in several different ways, interfering with cell division by mitosis. The damaged DNA elicits DNA repair mechanisms, which in turn activate apoptosis when repair proves impossible. Non-covalently bound drugs : 1. Minor groove binders- Minor groove binding drugs are usually crescent shaped, which complements the shape of the groove and facilitates binding by promoting van der Waals interactions. Additionally, these drugs can form hydrogen bonds to bases, typically to N3 of adenine and O2 of thymine. Most minor groove binding drugs bind to A/T rich sequences.
This preference in addition to the designed propensity for the electronegative pockets of AT sequences is probably due to better van der Waals contacts between the ligand and groove walls in this region, since A/T regions are narrower than G/C groove regions and also because of the steric hindrance in the latter, presented by the C2 amino group of the guanine base. However, a few synthetic polyamides like lexitropsins and imidazole-pyrrole polyamides have been designed which have specificity for G-C and C-G regions in the grooves. 2. Intercalators- These contain planar heterocyclic groups which stack between adjacent DNA base pairs. The complex, among other factors, is thought to be stabilized by - stacking interactions between the drug and DNA bases. Intercalators introduce strong structural perturbations in DNA.
Non-covalent binding is reversible and is typically preferred over covalent adduct formation keeping the drug metabolism and toxic side effects in mind. However, the high binding strength of covalent binders is a major advantage. Proteins are large molecules and bind quite strongly to the DNA, with binding constants in the nanomolar range. It has been difficult to achieve similar specificity and affinity using small non-covalent binders, and remains a major challenge to the design of drugs for DNA. Some DNA binders are listed in the following table Table 1. Drug, action and mode of binding for some DNA binding drugs. SNo 1 2 3 4 5 6 7 8 9 10 Drug Action Mode of Binding PDB Hoechst 33258 Antitumor Minor groove binding 264D Netropsin Antitumor, Antiviral Minor groove binding 121D Pentamidine Active against P. carinii Minor groove binding 1D64 Berenil Antitrypanosomal Minor groove binding 1D63 Guanyl bisfuramidine Active against P. carinii Minor groove binding 227D Netropsin Antitumor, Antiviral Minor groove binding 121D Distamycin Antitumor, Antiviral Minor groove binding 2DND SN7167 Antitumor, Antiviral Minor groove binding 328D SN6999 Active against P. falciparum Minor groove binding 144D Nogalamycin Antitumor Intercalation 182D
11 12 13 14 15
Menogaril Antitumor- Topoisomerase II poison Intercalation Mithramycin Anticancer antibiotic Minor groove binding Plicamycin Anticancer antibiotic Minor groove binding 1BP8 Chromomycin A3 Anticancer antibiotic Minor groove binding cis -Platin Anticancer antibiotic Covalent cross-linking
202D 146D 1EKH 1AU5
Forces involved in DNA-drug recognition: Understanding the forces involved in the binding of proteins or small molecules to DNA is of prime importance due to two major reasons. Firstly, the design of sequence specific drugs having requisite affinity for DNA requires a knowledge how the structure of the drug is related to the specificity/affinity of binding and what structural modifications could result in a drug with desired qualities. Secondly, identifying the forces/energetics involved in such processes is fundamental to unraveling the mystery of molecular recognition in general and DNA binding in particular. Some of the forces that are known to contribute to biomolecular recognition and also to DNA-drug binding are direct electrostatic interactions, direct van der Waals/packing interactions, complex hydration/dehydration contributions composed of hydrophobic component, solvation electrostatics, solvation van der Waals, ion effects and entropy terms. DNA-drug binding may be described in the following manner,
Consider DNA-drug binding in an aqueous environment. DNA is polyanionic in nature and the drug molecule is also often charged. The associated counterions lie near the charged groups and are also partially solvated. When binding occurs, it results in a displacement of solvent from the binding site on both the DNA and drug. Also, since there would be partial compensation of charges as the DNA and drug are oppositely charged, some counterions would be released into the bulk solvent and are solvated fully. Also, the binding process would be associated with some structural deformation/adaptation of the DNA as well as the drug molecule in order to accommodate each other. All these events are associated with some energetic gains/losses, the comprehensive estimation of which is a major challenge. We are attempting to understand the energetics of DNA-drug interaction by theoretically estimating the above contributions employing classical and statistical mechanical methods. Developing a theoretical protocol for detailed quantitative analysis of DNA-ligand binding in solution is a daunting task due to some major challenges. Simulations of DNA with solvent and the attendant counterion atmosphere require careful consideration to ensure system stability. Also, evolving a computationally efficient technique using statistical mechanical principles for quantitative estimates of binding free energies in large biomolecular systems is an equally challenging task. Our study is aimed at providing such a theoretical protocol for complementing experimental techniques and facilitating a minute study of the structure-energy relationships in DNA-drug complexes. Structural and conformational changes in the DNA and drug on binding in solution are associated with enthalpic and entropic contributions to the binding free energy, which can be theoretically estimated from ensembles of structures generated via simulations. The only drawback of this approach is the long time
taken for the simulations. The other terms, namely, electrostatics, van der Waals, hydrophobic component, rotational and translational entropy can be estimated from single structures. The web tool, PreDDICTA, estimates the components of DNA-drug binding free energy which can be calculated from a single structure, and correlates it with experimental binding free energy and Tm, thus providing a swift method for evaluation of potential lead candidates for researchers pursuing structure based drug design for DNA.
References and Further Reading 1. Neidle, S., Thurston, D.E. (2005) Chemical approaches to the discovery and development of cancer therapies Nat Rev Cancer, 5, 285-96. 2. Geierstanger, B.H., Wemmer, D.E. (1995) Complexes of the minor groove of DNA. Annu. Rev. Biophys. Biomol. Struct., 24, 463-493. 3. Chaires, J. B. (1998) Drug--DNA interactions. Curr. Opin. Struc. Biol., 8, 314320. 4. Neidle, S. (2001) DNA minor-groove recognition by small molecules. Nat. Prod. Rep., 18, 291-309. 5. Hurley, L. H. (2002) DNA and its associated processes as targets for cancer therapy. Nature Reviews Cancer, 2, 188-200. 6. Wemmer, D. E., Dervan, P. B. (1997) Targeting the minor groove of DNA. Curr. Opin. Struc. Biol., 7, 355-61. 7. Jones, S, van Heyningen, P, Berman, H. M., Thornton, J. M. (1999) Protein-DNA interactions: A structural analysis. J. Mol. Biol., 287, 877-896. 8. Jen-Jacobson, L. (1997) Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers, 44,153-180. 9. Janin, J. (1999) Wet and dry interfaces: the role of solvent in protein-protein and protein-DNA recognition. Structure Fold Des., 7, R277-279. 10. Turner, P. R., Denny, W. A. (2000) The genome as a drug target: sequence specific minor groove binding ligands Curr. Drug Targ., 1, 1-14. 11. Goodsell, D. S. (2001) Sequence recognition of DNA by lexitropsins. Curr. Med. Chem., 8, 509-516. 12. Reddy, B. S., Sondhi, S. M., Lown, J. W. (1999) Synthetic DNA minor groovebinding drugs. Pharmacol. Ther., 84, 1-111. 13. Dervan, P. B., Edelson, B. S. (2003) Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr. Opin. Struct. Biol., 13, 284-299. 14. Jayaram, B., Beveridge, D.L. (1996) Modeling DNA in aqueous solutions: theoretical and computer simulation studies on the ion atmosphere of DNA. Annu. Rev. Biophys. Biomol. Struct., 25, 367-394. 15. Auffinger, P., Westhof, E. (1998) Simulations of the molecular dynamics of nucleic acids. Curr. Opin. Struct. Biol., 8, 227236. STRUCTURAL BIOLOGY ASSIGNMENT DNA-DRUG INTERACTION
MADE BY: Name: VASU R SAH No.: 38038 Semester: Vth (AIB) DNA Drug Interaction Deoxyribonucleic acid, DNA, is a molecule of great biological significance. The total DNA content of a cell is termed the Genome. The Genome is unique to an organism, and is the information bank governing all life processes of the organism, DNA being the form in which this information is stored. Stretches of DNA called genes have the extremely important function of coding for proteins. The function of the rest of the genome, loosely termed as non-gene regions, is not very clearly known. Fig.1 the DNA molecule DNA has two main functions: 1. Transcription: Information is retrieved from the DNA by ribonucleic acid, RNA, and utilized to synthesize proteins in the body. Proteins are involved in all body processes and play many roles. e.g. as hormones, enzymes, carriers, structural proteins, receptors, regulators etc. 2. Replication: DNA is responsible for its own regeneration, i.e., DNA self replicates. DNA as carrier of genetic information is a major target for drug interaction because of the ability to interfere with transcription (gene expression and protein synthesis) and DNA replication, a major step in cell growth and division. The latter is central for tumorigenesis and pathogenesis. There are three principally different ways of drug binding: First, through control of transcription factors and polymerases. Here, the drugs interact with the proteins that bind to DNA. Second, through RNA binding to DNA double helices to form nucleic acid triple helical structures or RNA hybridization (sequence specific binding) to exposed DNA single strand regions forming DNA-RNA hybrids that may interfere with transcriptional activity. Third, small aromatic ligand molecules that bind to DNA double helical structures by (i) intercalating between stacked base pairs thereby distorting the DNA backbone conformation and interfering with DNA-protein interaction or (ii) the minor groove binders. The latter cause little distortion of the DNA backbone. Both work through non-covalent interaction. The small ligand drug approach offers a simple solution. The synthesis and screening of synthetic compounds that do not exist in nature, work much like Roll Section: R
pharmacological ligand for cell surface receptors in excitable tissue, and appear to be more readily delivered to cellular targets than large RNA or protein ligands. The lack of sequence specificity for intercalating molecules, however, does not allow to target specific genes, but rather certain cellular states or physiological and pathological conditions, like rapid cell growth and division that can be selectively suppressed as compared to non growing or slowly growing healthy tissue. DNA is present in the body in the form of a double helix, where each strand is composed of a combination of four nucleotides, adenine (A), thymine (T), guanine (G) and cytosine (C). Within a strand these nucleotides are connected via phosphodiester linkages. The two strands are held together primarily via Watson Crick hydrogen bonds where A forms two hydrogen bonds with T and C forms three hydrogen bonds with G (Figure2). Fig.2 Watson Crick Base pairing, A-T and G-C base pairing Specific recognition of DNA sequences by proteins/ small molecules is achieved via the combination of hydrogen bond acceptor/donor sites available on the major groove or minor groove. e.g. the A-T base pair offers a hydrogen bond acceptor, N7, a donor N6, and an acceptor, O4 on the major groove side. DNA-Drug Interaction : Transcription and replication are vital to cell survival and proliferation as well as for smooth functioning of all body processes. DNA starts transcribing or replicating only when it receives a signal, which is often in the form of a regulatory protein binding to a particular region of the DNA. Thus, if the binding specificity and strength of this regulatory protein can be mimicked by a small molecule, then DNA function can be artificially modulated, inhibited or activated by binding this molecule instead of the protein. Thus, this synthetic/natural small molecule can act as a drug when activation or inhibition of DNA function is required to cure or control a disease (Table 1). DNA activation would produce more quantities of the required protein, or could induce DNA replication; depending on which site the drug is targeted. DNA inhibition would restrict protein synthesis, or replication, and could induce cell death. Though both these actions are possible, mostly DNA is targeted in an inhibitory mode, to destroy cells for antitumor and antibiotic action. Drugs bind to DNA both covalently as well as non-covalently. Covalent binding in DNA is irreversible and invariably leads to complete inhibition of DNA processes and subsequent cell death. Cis-platin (cisdiamminedichloroplatinum) is a famous covalent binder used as an anticancer drug, and makes an intra/interstrand cross-link via the chloro groups with the nitrogens on the DNA bases.
Non-covalently bound drugs mostly fall under the following two classes: 1. Minor groove bindersAdditionally, these drugs can form hydrogen bonds to bases, typically to N3 of adenine and O2 of thymine. Most minor groove binding drugs bind to A/T rich sequences.
This preference in addition to the designed propensity for the electronegative pockets of AT sequences is probably due to better van der Waals contacts between the ligand and groove walls in this region, since A/T regions are narrower than G/C groove regions and also because of the steric hindrance in the latter, presented by the C2 amino group of the guanine base. However, a few synthetic polyamides like lexitropsins and imidazole-pyrrole polyamides have been designed which have specificity for G-C and C-G regions in the grooves. 2. IntercalatorsThese contain planar heterocyclic groups which stack between adjacent DNA base pairs. The complex, among other factors, is thought to be stabilized by - stacking interactions between the drug and DNA bases. Intercalators introduce strong structural perturbations in DNA.
Non-covalent binding is reversible and is typically preferred over covalent adduct formation keeping the drug metabolism and toxic side effects in mind. However, the high binding strength of covalent binders is a major advantage. Proteins are large molecules and bind quite strongly to the DNA, with binding constants in the nanomolar range. It has been difficult to achieve similar specificity and affinity using small non-covalent binders, and remains a major challenge to the design of drugs for DNA. Modeling DNA-ligand interaction of intercalating ligands: The following properties have been identified as important for the successful modeling of ligand-DNA interaction: Degrees of freedom Role of base pair sequence Counter ion effects Role of solvent ligand-receptor binding Degrees of freedom this problem is analogous to that of protein ligand interaction. The major requirement for intercalating agents is the planar aromatic ring structure. This structure fits between to adjacent base pair planes and can have some, although much restricted, rotational freedom within the plane of the ring. The ligand itself may have flexibility of structural parts outside the DNA binding site and may contain more than one intercalating sidechain: The structure of the antibiotic triostin A shows the presence of two quinoxaline (groups to the right; double aromatic rings) units linked through a cyclic peptide structure (center left) which is stabilized at its center by a cystein pair (disulfhydril covalent bond).
Fig. Chemical structure of triostatin A The space filled side view indicates how the two quinoxaline rings are positioned by the linker peptide in co-planar fashion suitable for intercalating with DNA base pairs. As a rule, the more intercalating sidechains are linked within a single ligand structure, the stronger the expected binding affinity. Triostatin A belongs to a family of antibiotics which are characterized by crosslinked octapeptide rings bearing two quinoxaline chromophores. Since the spacing
between the chromophores is 3.5A, the intercalation process sandwiches two base pairs between the two quinoxalines. This phenomenon is called bis-intercalation and has first been described for echinomycin by showing that bis-intercalating drugs cause twice the DNA helix extension and unwinding seen as compared to single intercalating molecule like ethidium. The latter is a chromophore which is activated by UV light and is used by molecule biologists to label nucleic acids in gel electrophoresis or ion gradient centrifugation. Role of base pair sequence Experimental evidence suggests that base pair sequence does not play a large role on the specific mature of most intercalating complexes. As the structure of triostatin A suggests, however, the linker peptide structure may well promote specific interaction with the DNA surface. The major group specific readout sequence of H-bond donor and acceptor could be involved in triostatin A binding. The figure graphically shows the direct readout of the DNA base sequence on a double helical structure. Fig. Overlap of AT and GC base pairs The following characteristics of non covalent bond formation are associated with the binding sites indicated above: binding site GC base pair AT base pair W1 H-bond acceptor H-bond acceptor W2 blank blank W3 H-bond acceptor H-bond donor W3' blank blank W2' H-bond donor H-bond acceptor W1' C-H weak hydrophobic CH3, strong hydrophobic While the interaction on the major groove side is distinct for the direction of the base pair (e.g. AT vs TA), there is no directionality at the minor groove side. The molecular basis of specific recognition between echinomycin and DNA is due to the hydrogen bonding between the ligand alanine carbonyl groups and the 2-amino group of guanine. This is consistent with the observation that the preferred binding site is the sequence CG. Counter ion effect DNA is a negatively charged polyanion attracting counter ions, positively charged Na+, or Ca++ and Mg++ ions as well as basic residues of proteins. The presence of small counter ion affect drug binding, since the counter ions can screen and shield the negative backbone surface allowing non electrolytes as well as positively charged ligand to interact more strongly with the DNA target. High ionic strength, however, reduces non covalent interaction mediated by hydrogen bonds and electrostatic interactions. Role of solvent ligand-receptor binding There are three general classes of interactions that must be considered in solvated ligand-receptor binding : (a) ligand solvent interaction (e.g. hydration shell), (b) receptor solvent interaction, and (c) ligand-DNA complex with solvent interaction. The three classes basically describe the sequence of events of free ligand interacting with its receptor and the change in overall solvent interaction before and after binding. We have seen that the hydrophobic effect is completely described by this system and the contribution of the entropy of free bulk water is the major driving force of hydrophobic ligand receptor interaction. This type of interaction is found in intercalating substrates because the hydrophobic, aromatic sidechains interactive favorably with the aromatic environment of the base pair stacking. The total amount of surface bound water is reduced in the after complex formation. Rational for drug design
When a compound intercalates into nucleic acids, there are changes which occur in both the DNA and the compound during complex formation that can be used to study the ligand DNA interaction. The binding is of course an equilibrium process because no covalent bond formation is involved. The binding constant can be determined by measuring the free and DNA bound form of the ligand. Since many of the intercalating substrates are aromatic chromophores, this can be done spectroscopically. Also, DNA double helix structures are found to be more stable with intercalating agents present and show a reduced heat denaturation. Correlating these biophysical parameters with cytotoxicity is used to support the antitumor activity of these drugs as based on their ability to intercalate in DNA double helical structures. Improvement of anticancer drugs based on intercalating activity is not only focussed on DNA-ligand interaction, but also on tissue distribution and toxic side effects on the heart (cardiac toxicity) due to redox reduction of the aromatic rings and subsequent free radical formation. Free radical species are thought to induce destructive cellular events such as enzyme inactivation, DNA strand cleavage and membrane lipid peroxidation. Modeling DNA-ligand interaction of minor groove binders: Hairpin minor grove binding molecules have been identified and synthesized that bind to GC reach nucleotide sequences. Hairpin polyamides are linked systems that exploit a set of simple recognition rules for DNA base pairs through specific orientation of imidazole (Im) and pyrrole (Py) rings. The hairpin polyamides originated from the discovery of the three-ring Im-Py-Py molecule that bound to minor groove DNA as an antiparallel side by side dimer. Fig. Structure of hairpin ligand (right) on DNA minor groove (left)
The compound was found to recognize GC base pairs. Solid phase synthesis of polyamides of variable length has produced efficient ligands, e.g. the eight ring hairpin polyamide ImPyPyPy-g-ImPyPyPy-b-Dp (Dp dimethylamino propylamide) shown in the figure above. This small synthetic molecule has an binding constant in the order of 0.03nM. The optimal goal of polyamide ligand design has been reached with finding structures able to recognize DNA sequences of specific genes. The structure shown above inhibits the expression of 5S RNA in fibroblast cells (skin cancer cells) by interfering with the transcription factor IIIA-binding site. A new strategy of rational drug design exploits the combination of polyamides with bis-intercalating structures. WP631 is a dimeric analog of the clinically proven anthracycline antibiotic daunorobuicin. Fig. Structure of WP631 This new synthetic compound shows an affinity of 10pM and also showed to be resistant against multidrug resistance mechanisms often encountered in antitumor therapy. Multidrug resistance is a phenomenon where small aromatic compounds are efficiently expelled from the cell by cell membrane transport proteins commonly referred to as ABC transporters (or ATP Binding Cassette proteins). Drugs that form covalent bonds with DNA targets Drugs that interfere with DNA function by chemically modifying specific nucleotides are Mitomycin C, Cisplatin, and Anthramycin. Mitomycin C is a well characterized antitumor antibiotic which forms a covalent interaction with DNA after reductive activation. The activated antibiotic forms a cross-linking structure between guanine bases on adjacent strands of DNA thereby inhibiting single strand formation (this is essential for mRNA transcription and DNA replication).
Anthramycin is an antitumor antibiotic which bind covalently to N-2 of guanine located in the minor groove of DNA. Anthramycin has a preference of purine-Gpurine sequences (purines are adenine and guanine) with bonding to the middle G. Cisplatin is a transition metal complex cis-diamine-dichloro-platinum and clinically used as anticancer drug. The effect of the drug is due to the ability to platinate the N-7 of guanine on the major groove site of DNA double helix. This chemical modification of platinum atom cross-links two adjacent guanines on the same DNA strand interfering with the mobility of DNA polymerases. Forces involved in DNA-drug recognition: Understanding the forces involved in the binding of proteins or small molecules to DNA is of prime importance due to two major reasons. Firstly, the design of sequence specific drugs having requisite affinity for DNA requires a knowledge how the structure of the drug is related to the specificity/affinity of binding and what structural modifications could result in a drug with desired qualities. Secondly, identifying the forces/energetics involved in such processes is fundamental to unraveling the mystery of molecular recognition in general and DNA binding in particular. Some of the forces that are known to contribute to biomolecular recognition and also to DNA-drug binding are direct electrostatic interactions, direct van der Waals/packing interactions, complex hydration/dehydration contributions composed of hydrophobic component, solvation electrostatics, solvation van der Waals, ion effects and entropy terms. DNA-drug binding may be described in the following manner:
Consider DNA-drug binding in an aqueous environment. DNA is polyanionic in nature and the drug molecule is also often charged. The associated counterions lie near the charged groups and are also partially solvated. When binding occurs, it results in a displacement of solvent from the binding site on both the DNA and drug. Also, since there would be partial compensation of charges as the DNA and drug are oppositely charged, some counterions would be released into the bulk solvent and are solvated fully. Also, the binding process would be associated with some structural deformation/adaptation of the DNA as well as the drug molecule in order to accommodate each other. All these events are associated with some energetic gains/losses, the comprehensive estimation of which is a major challenge. We are attempting to understand the energetics of DNA-drug interaction by theoretically estimating the above contributions employing classical and statistical mechanical methods. Developing a theoretical protocol for detailed quantitative analysis of DNA-ligand binding in solution is a daunting task due to some major challenges. Simulations of DNA with solvent and the attendant counterion atmosphere require careful consideration to ensure system stability. Also, evolving a computationally efficient technique using statistical mechanical principles for quantitative estimates of binding free energies in large biomolecular systems is an equally challenging task. Our study is aimed at providing such a theoretical protocol for complementing experimental techniques and facilitating a minute study of the structure-energy relationships in DNA-drug complexes. Structural and conformational changes in the DNA and drug on binding in solution are associated with enthalpic and entropic contributions to the binding free energy, which can be theoretically estimated from ensembles of structures generated via simulations. The only drawback of this approach is the long time
taken for the simulations. The other terms, namely, electrostatics, van der Waals, hydrophobic component, rotational and translational entropy can be estimated from single structures. The web tool, PreDDICTA, estimates the components of DNA-drug binding free energy which can be calculated from a single structure, and correlates it with experimental binding free energy and Tm, thus providing a swift method for evaluation of potential lead candidates for researchers pursuing structure based drug design for DNA. Chapter-21 Technique to study protein Introduction: Proteins are fundamental components of all living cells. They exhibit an enormous amount of chemical and structural diversity, enabling them to carry out an extraordinarily diverse range of biological functions. Scientists know that the critical feature of a protein is its ability to adopt the right shape for carrying out a particular function. But sometimes a protein twists into the wrong shape or has a missing part, preventing it from doing its job. Many diseases, such as Alzheimer's and "mad cow", are now known to result from proteins that have adopted an incorrect structure. Identifying a protein's shape, or structure, is a key to understanding its biological function and its role in health and disease. Illuminating a protein's structure also paves the way for the development of new agents and devices to treat a disease. Yet solving the structure of a protein is no easy feat. It often takes scientists working in the laboratory months, sometimes years, to experimentally determine a single structure. Therefore, scientists have begun to turn toward computers to help predict the structure of a protein based on its sequence. The challenge lies in developing methods for accurately and reliably understanding this intricate relationship.
Determination of Protein Structure Traditionally, a protein's structure was determined using one of two techniques: 1.X-ray Crystallography. 2.Nuclear Magnetic Resonance (NMR) Spectroscopy. X-ray Crystallography . The basic building block of a crystal is called a unit cell. Each unit cell contains exactly one unique set of the crystal's components, the smallest possible set that is fully representative of the crystal. Crystals of a complex molecule, like a protein, produce a complex pattern of X-ray diffraction, or scattering of X-rays. When the crystal is placed in an X-ray beam, all of the unit cells present the same face to the beam; therefore, many molecules are in the same orientation with respect to the incoming X-rays. The X-ray beam enters the crystal and a number of smaller beams emerge: each one in a different direction, each one with a different intensity. If an X-ray detector, such as a piece of film, is placed on the opposite side of the crystal from the X-ray source, each diffracted ray, called a reflection, will produce a spot on the film. However, because only a few reflections can be detected with any one orientation of the crystal, an important component of any X-ray diffraction instrument is a device for accurately setting and changing the orientation of the crystal. The set of diffracted, emerging beams contains information about the underlying crystal structure.
Nuclear Magnetic Resonance(NMR)Spectroscopy The basic phenomenon of NMR spectroscopy was discovered in 1945. In this technique, a sample is immersed in a magnetic field and bombarded with radio waves. These radio waves encourage the nuclei of the molecule to resonate, or spin. As the positively charged nucleus spins, the moving charge creates what is called a magnetic moment. The thermal motion of the moleculethe movement of the molecule associated with the temperature of the materialfurther creates a torque, or twisting force, that makes the magnetic moment "wobble" like a child's top. When the radio waves hit the spinning nuclei, they tilt even more, sometimes flipping over. These resonating nuclei emit a unique signal that is then picked up on a special radio receiver and translated using a decoder. This decoder is called the Fourier Transform algorithm, a complex equation that translates the language of the nuclei into something a scientist can understand. By measuring the frequencies at which different nuclei flip, scientists can determine molecular structure, as well as many other interesting properties of the molecule. In the past 10 years, NMR has proven to be a powerful alternative to X-ray crystallography for the determination of molecular structure. NMR has the advantage over crystallographic techniques in that experiments are performed in solution as opposed to a crystal lattice. However, the principles that make NMR possible tend to make this technique very time consuming and limit the application to small- and medium-sized molecules. Strategy behind NMR Nuclear magnet resonance obtains the same high resolution using a very different strategy. NMR measures the distances between atomic nuclei, rather than the electron density in a molecule. With NMR, a strong, high frequency magnetic field stimulates atomic nuclei of the isotopes H-1, D-2, C-13, or N-15 (they have a magnetic spin) and measures the frequency of the magnetic field of the atomic nuclei during its oscillation period back to the initial state. The important step is to determine which resonance comes from which spin. The distance and type of neighboring nuclei determines the resonance frequency of the stimulated atomic nuclei. This dependence on next neighbors known as chemical shift (or spin-spin coupling constant) and reflects the local electronic environment and the information contained in 1-D NMR spectra. For proteins, NMR usually measures the spin of protons. The following reasons make the H-1 NMR spectroscopy the method of choice for biological macromolecules: - H is present at many sites in proteins, nucleic acids, and polysaccharides - H has a high abundance for each site - H nuclei is the most sensitive to detect
1-D spectra contain the information about all the chemical shifts of all the H in the protein. The frequency resolution is often not enough to distinguish individual chemical shifts. 2-D NMR solves these problems by containing information about the relative position of H in molecular structures. 2-D NMR spectra contain information about interaction between H that is covalently linked through one or two other atoms (COSY or correlation spectroscopy). Alternatively, pairs of H that can be close in space, even if they are from residues that are not close in sequence (NOE spectra, or Nuclear Overhauser Effect). A complete structure can thus be calculated by sequentially assigning cross peak correlations in 2-D spectra. Currently, the size limit for proteins amenable to NMR solution structure analysis is about 200 amino acids. An important feature of the identification of cross peaks is that regular patterns can be recognized that stem from secondary structure elements such as alpha helices and parallel or anti-
parallel beta sheets because they contain typical hydrogen bonding networks. NMR also requires the knowledge of the amino acid sequence, but the protein does not have to be in an ordered crystal, yet high concentrations of solubilized protein must be available (NMR structures are therefore also called solution structures). In biopolymers, the primary structure (sequence) logically breaks up the molecule into groups of coupled spins normally one or two groups per residue. This is true not only for proteins, but also for nucleic acids and polysaccharides. Fig. Observed NOEs in antiparallel and parallel sheets
Determination of membrane protein structure by rotational resonance NMR: bacteriorhodopsin. Rotationally resonant magnetization exchange, a new nuclear magnetic resonance (NMR) technique for measuring internuclear distances between like spins in solids, was used to determine the distance between the C-8 and C-18 carbons of retinal in two model compounds and in the membrane protein bacteriorhodopsin. Magnetization transfer between inequivalent spins with an isotropic shift separation, delta, is driven by magic angle spinning at a speed omega r that matches the rotational resonance condition delta = n omega r, where n is a small integer. The distances measured in this way for both the 6-s-cis- and 6-s-trans-retinoic acid model compounds agreed well with crystallographically known distances. In bacteriorhodopsin the exchange trajectory between C-8 and C-18 was in good agreement with the internuclear distance for a 6-s-trans configuration [4.2 angstroms (A)] and inconsistent with that for a 6-s-cis configuration (3.1 A). The results illustrate that rotational resonance can be used for structural studies in membrane proteins and in other situations where diffraction and solution NMR techniques yield limited information. Structure of bacteriorhodopsin:
General: Current strategies for determining the structures of membrane proteins in lipid environments by NMR spectroscopy rely on the anisotropy of nuclear spin interactions, which are experimentally accessible through experiments performed on weakly and completely aligned samples. Importantly, the anisotropy of nuclear spin interactions results in a mapping of structure to the resonance frequencies and splittings observed in NMR spectra. Distinctive wheel-like patterns are observed in two-dimensional 1H-15N heteronuclear dipolar/15N chemical shift PISEMA (polarization inversion spin-exchange at the magic angle) spectra of helical membrane proteins in highly aligned lipid bilayer samples. One-dimensional dipolar waves are an extension of two-dimensional PISA (polarity index slant angle) wheels that map protein structures in NMR spectra of both weakly and completely aligned samples. Dipolar waves describe the periodic wave-like variations of the magnitudes of the heteronuclear dipolar couplings as a function of residue number in the absence of chemical shift effects. Since weakly aligned samples of proteins display these same effects, primarily as residual dipolar couplings, in solution NMR spectra, this represents a convergence of solid-state and solution NMR approaches to structure determination
X-ray crystallography and NMR are complementary techniques.
NMR spectroscopy X-ray crystallography short time scale, protein folding long time scale, static structure solution, purity single crystal, purity < 20kD, domain any size, domain, complex functional active site active or inactive domains domains atomic nuclei, chemical bonds electron density resolution limit 2-3.5 resolution limit 2-3.5 primary structure must be known primary structure must be know (except if resolution is 2 or better for every single residue)
References: 1. Science 15 February 1991: Vol. 251. no. 4995, pp. 783 - 786 DOI: 10.1126/science.1990439 ARTICLE Determination of membrane protein structure by rotational resonance NMR: bacteriorhodopsin F Creuzet, A McDermott, R Gebhard, K van der Hoef, MB Spijker-Assink, J Herzfeld, J Lugtenburg, MH Levitt, and RG Griffin. 2. Wikipedia.
3. Biochemistry by Lehninger, Chapter no.4: The three dimensional structure of Proteins, page no.116.
Chapter-22 Enzymology of DNA folding and unfolding WHAT IS DNA???
Deoxyribonucleic acid (DNA) is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms. The main role of DNA molecules is the long-term storage of information and DNA is often compared to a set of blueprints, since it contains the instructions needed to construct other components of cells, such as proteins and RNA molecules. The DNA segments that carry this genetic information are called genes, but other DNA sequences have structural purposes, or are involved in regulating the use of this genetic information. Chemically, DNA is a long polymer of simple units called nucleotides, with a backbone made of sugars and phosphate groups joined by ester bonds. Attached to each sugar is one of four types of molecules called bases. It is the sequence of these four bases along the backbone that encodes information. This information is read using the genetic code, which specifies the sequence of the amino acids within proteins. The code is read by copying stretches of DNA into the related
nucleic acid RNA, in a process called transcription. Most of these RNA molecules are used to synthesize proteins, but others are used directly in structures such as ribosomes and spliceosomes. Within cells, DNA is organized into structures called chromosomes and the set of chromosomes within a cell make up a genome. These chromosomes are duplicated before cells divide, in a process called DNA replication. Eukaryotic organisms such as animals, plants, and fungi store their DNA inside the cell nucleus, while in prokaryotes such as bacteria it is found in the cell's cytoplasm. Within the chromosomes, chromatin proteins such as histones compact and organize DNA, which helps control its interactions with other proteins and thereby control which genes are transcribed.
DNA STRUCTURE
DNA is a long polymer made from repeating units called nucleotides.[1][2] The DNA chain is 22 to 26 ngstrms wide (2.2 to 2.6 nanometres), and one nucleotide unit is 3.3 ngstroms (0.33 nanometres) long.[3] Although each individual repeating unit is very small, DNA polymers can be enormous molecules containing millions of nucleotides. For instance, the largest human chromosome, chromosome number 1, is 220 million base pairs long.[4] In living organisms, DNA does not usually exist as a single molecule, but instead as a tightly-associated pair of molecules.[5][6] These two long strands entwine like vines, in the shape of a double helix. The nucleotide repeats contain both the segment of the backbone of the molecule, which holds the chain together, and a base, which interacts with the other DNA strand in the helix. In general, a base linked to a sugar is called a nucleoside and a base linked to a sugar and one or more phosphate groups is called a nucleotide. If multiple nucleotides are linked together, as in DNA, this polymer is referred to as a polynucleotide.[7] The backbone of the DNA strand is made from alternating phosphate and sugar residues.[8] The sugar in DNA is 2-deoxyribose, which is a pentose (five carbon) sugar. The sugars are joined together by phosphate groups that form phosphodiester bonds between the third and fifth carbon atoms of adjacent sugar rings. These asymmetric bonds mean a strand of DNA has a direction. In a double helix the direction of the nucleotides in one strand is opposite to their direction in the other strand. This arrangement of DNA strands is called antiparallel. The asymmetric ends of DNA strands are referred to as the 5 (five prime) and 3 (three prime) ends. One of the major differences between DNA and RNA is the sugar, with 2-deoxyribose being replaced by the alternative pentose sugar ribose in RNA.[6] The DNA double helix is stabilized by hydrogen bonds between the bases attached to the two strands. The four bases found in DNA are adenine (abbreviated A), cytosine (C), guanine (G) and thymine (T). These four bases are shown below and are attached to the sugar/phosphate to form the complete nucleotide, as shown for adenosine monophosphate. These bases are classified into two types; adenine and guanine are fused five- and six-membered heterocyclic compounds called purines, while cytosine and thymine are six-membered rings called pyrimidines.[6] A fifth pyrimidine base, called uracil (U), usually takes the place of thymine in RNA and differs from thymine by lacking
a methyl group on its ring. Uracil is not usually found in DNA, occurring only as a breakdown product of cytosine, but a very rare exception to this rule is a bacterial virus called PBS1 that contains uracil in its DNA.[9] In contrast, following synthesis of certain RNA molecules, a significant number of the uracils are converted to thymines by the enzymatic addition of the missing methyl group. This occurs mostly on structural and enzymatic RNAs like transfer RNAs and ribosomal RNA.[ 0] Major and minor grooves Animation of the structure of a section of DNA. The bases lie horizontally between the two spiraling strands. Large version[11] The double helix is a right-handed spiral. As the DNA strands wind around each other, they leave gaps between each set of phosphate backbones, revealing the sides of the bases inside (see animation). There are two of these grooves twisting around the surface of the double helix: one groove, the major groove, is 22 wide and the other, the minor groove, is 12 wide.[12] The narrowness of the minor groove means that the edges of the bases are more accessible in the major groove. As a result, proteins like transcription factors that can bind to specific sequences in double-stranded DNA usually make contacts to the sides of the bases exposed in the major groove.[13]
Replication Cell division is essential for an organism to grow, but when a cell divides it must replicate the DNA in its genome so that the two daughter cells have the same genetic information as their parent. The double-stranded structure of DNA provides a simple mechanism for DNA replication. Here, the two strands are separated and then each strand's complementary DNA sequence is recreated by an enzyme called DNA polymerase. This enzyme makes the complementary strand by finding the correct base through complementary base pairing, and bonding it onto the original strand. As DNA polymerases can only extend a DNA strand in a 5 to 3 direction, different mechanisms are used to copy the antiparallel strands of the double helix.[71] In this way, the base on the old strand dictates which base appears on the new strand, and the cell ends up with a perfect copy of its DNA
MECHANISM OF DENATURATION AND RENATURATION OF DNA DNA DENATURATION :
The two DNA strands, which form the double helix, are in opposite orientations (one runs in 5'-3'direction, the complementary in 3'-5'direction) and are connected by hydrogen bonds (see DNA structure). The rupture of these hydrogen bonds causes the DNA to separate or to "denature". Inside the cell, a partial separation of the DNA strands is necessary for replication and transcription and is caused by several proteins that use ATP. But in vitro the rupture of the hydrogen bonds can be caused by heat, alcaline conditions or chemical compounds The denaturation by heat is complete at 90C. Concerning alcalic conditions, a pH above 11.3 is able to denature DNA. In both cases, the process of denaturation is reversible. In the moment that the denaturating agent disappears, the DNA renaturalizes ( reassociates, reanneals) and it gets back to its native structures of a double helix. The temperature at which half of the molecules are denatured is called Tm (melting temperature). The Tm varies among organisms depending on their porcentage of G+C. A=T rich zones melt at lower temperatures than G=C rich zones, whereas GC rich zones tends to have a higher Tm. The content of G+C can therefore be calculated from the Tm value of the DNA. Also in biological systems, replication and transcription begins in the A=T rich regions. Denaturation at an elavated pH > 11 is caused by a rupture of the hydrogen bonds between the complementary bases. The alcalic conditions change the charges of the many side groups that are involved in the non-covalent binding between the bases. Distilled water can also cause some degree of denaturation. It inhibits the neutralisation of the phosphate groups by salts like magnesium or sodium. The negative charge of the phopsphate group causes a repulsion and therefore a physical separation of the DNA strands. Chemical compounds like urea or formamide denature the DNA by directly reacting with the bases, thereby preventing normal base-pairing. The Tm is reduced proportionally to the concentration of denaturating chemical agents. If we decrease the concentration of either urea or formamide, DNA will renature again. Urea is used in denaturing gels to sequence DNA. Formaldehyde causes an irreversible denaturation, because it builds covalent bonds with the NH2 groups of the bases, thus preventing base-pairing. It can react with the DNA at room temperatures, because even in native DNA the bases are in a dynamic equilibrium of pairing and unpairing in discrete regions, called the respiration or palpitation of the DNA. It is thought that this phenomenon facilitates the access of regulatory proteins to the inner part of the DNA molecule. If DNA is to be kept in a denatured state, it needs to be transferred immediately from 100C to ice for fast cooling. This will prevent it from forming again the hydrogen bonds in an ordered manner. The denaturation of the DNA can be observed by a change of light absorption at a wavelength of 260 nm. Single stranded DNA has an appr. 40% higher absorption than double stranded DNA, since the heterocyclic rings of the bases absorb more light if they are not piled up or connected by hydrogen bonds insight the double helix. This phenomenon is called "hipercromicity". DNA RENATURATION :
The process of renaturation, observable as a change in light absorption, is a very valuable mechanism to evaluate the genetic relation between organisms. It shows
the degree of similarity, correspondance and divergance between DNAs of different origin. A DNA that contains a high quantity of repetitive sequences (satellite sequences) will renaturate much faster than DNA that is mainly single sequences. Mice DNA for example renatures faster that bacterial DNA, even so it is 30 time bigger, because it contains many repetitive sequences. Shorter DNA molecules reassociate faster than longer molecules. In long molecules, many small fragments appear as a result of shering forces and the possibility that the complementary segments "find each other" is lower. This fact is valuable for example in compairing phage- to bacterial DNA. The renaturation occurs is two steps: 1.Two complementary sequences collide by incidence and they combine forming a helix structure. 2. The Base-pairing is extended over the whole strand in form of a "zipper". The process of renaturation is also called hybridization, and it can happen with its original complementary strand, but also with another sequence that has high homology but not complete. Hybridization is the basis of many molecular techniques studied in This course. What is Denaturation of DNA ?? As DNA is heated, it reaches a temperature where the strands seperate (DNA melts). The base pairs are separated as the H-bonds between them are broken and the strand unwind. This results in single stranded DNA. It is possible to follow this process in a spectrophotometer, by observing the change in absorbance at 260nm. Unstacked bases (random orientation) absorb more light than neatly stacked (oriented) base-pairs. This results in a melting curve. The temperature at which DNA is half unfolded is called the melting temperature. Tm is a measure of the stability of DS-DNA under a given set of conditions. Stability, and therefore Tm, is affected by.... Base Composition - higher the GC content, the higher the Tm. Ionic Strength - as the ionic strength increases, so does Tm. Double helical DNA is stabilised by cations. Divalent cations (eg Mg2+) are more effective than monovalent cations (<NA+ or K+). Organic Solvents - formamide for instance lowers the Tm by weakening the hydrophobic interactions.
What is Reannealing of DNA ?? If melted DNA is cooled slowly, complementary strands will pair up again. This process is called "annealing". Only complementary strands can anneal. This is an important feature of DNA which is utilised in the laboratory when carrying out DNA hybridisation. Perfect annealing requires the perfect matching of base pairs, although mismatching occurs quite often. This gives less stable DNA, and can be monitored by watching the Tm. If the Tm lowers by 1 o, then 1% mismatch has occurred. The conditions of annealing, salt concentration and temperature, can determine the amount of mismatch that occurs. But it must be said that not all mismatching is bad. If, for instance, a gene was found in a rat, and researchers needed to know if a similiar gene was found in humans, then they would need to allow some mismatch, since the genetic sequences are unlikely to be exactly the same. "Stringency" is the term given to the conditions of annealing which control the
extent of mismatching allowed. If the stringency of hybridisation is high, there is little or no mismatch with 95-100% of base-pairs matched correctly. At low stringency, only 40-50% of base-pairs are correctly repaired. Renaturation of DNA by a Saccharomyces cerevisiae Protein That Catalyzes Homologous Pairing and Strand Exchange* A protein from mitotic Saccharomyces cerevisiae cells that catalyzes homologous pairing and stranedx change was analyzed for the ability to catalyze other related reactions. The proteiwna s capable of renaturing complementary single-strandedD NA as evidenced by S1 nuclease assays and analysis of the reaction products by agarose gel electrophoresis and electron microscopy. Incubation of the yeast protewini th complementary single-strandedD NA resulted in the rapid formation ofl arge aggregates which did noetn ter agarose gels. These aggregates contained many branched structures consisting of both single-stranded and doublestranded DNA. These reactions required stoichiometric amounts of protein but shonwoe Ad TP requirement. The protein formed stable complexesw ith both single-stranded and double-stranded DNAs,h oeing a higher affinity for single-stranded DNA. The binding to single-stranded DNA resulted in the formation of large protein:DNA aggregates. These aggregates were also formed in strand-exchange reactions and contained both substrate and product DNAs. Trheessuel ts demonstrate that the S. cerevisiae strand-exchange protein shares additional properties with the Escherichia coli recA protein which, by analogy, gives further indication that it might be implicated in homologous recombination
Mechanism of DNA denaturation in the presence of manganese ions
The unwinding of DNA strands in the presence of small concentrations of Mn2+ ions (2 10-4-4 10-4M) has been studied. The process of unwinding is nonequilibrium; the DNA strands are gradually unwound at a constant temperature corresponding to the beginning of the melting curve. There is no true renaturation in the partially melted DNA. It is shown in the paper that these effects are due to the aggregation of the unwound DNA regions. The Mn2+ ions are responsible for the binding of the unwound strands. The aggregation precludes renaturation, shifts the equilibrium towards the melted state, and causes slow unwinding at a constant temperature. The binding of denaturated regions seems to occur through the guanines
ADVANTAGES OF DENATURATION
1.PCR MELTING PROFILE(PCR MP)
The performance and convenience of a PCR melting profile (PCR MP) technique based on using low denaturation temperatures during ligation mediated PCR (LM PCR) of bacterial DNA is shown. We found that PCR MP technique is a rapid method that offers good discriminatory power, excellent reproducibility and may be applied for epidemiological studies. Results from strain genotyping illustrate that PCR MP is useful for the study of intraspecific genetic relatedness of strains and is as effective in discriminating closely related strains as the PFGE method, which is currently considered to be the gold standard for epidemiological studies. The usefulness of the PCR MP for molecular typing was shown for clinical strains of Escherichia coli, Enterococcus faecium VRE and Stapylococcus aureus. 2.HYBRIDIZATION OF DENATURED RNA AND SMALL DNA FRAGMENTS TRANSFERRED TO NITROCELLULOSE A simple and rapid method for transferring RNA from agarose gels to nitrocellulose paper for blot hybridization has been developed. Poly(A)+ and ribosomal RNAs transfer efficiently to nitrocellulose paper in high salt (3 M NaCl/0.3 M trisodium citrate) after denaturation with glyoxal and 50% (vol/vol) dimethyl sulfoxide. RNA also binds to nitrocellulose after treatment with methylmercuric hydroxide. The method is sensitive: about 50 pg of specific mRNA per band is readily detectable after hybridization with high specific activity probes (10(8) cpm/microgram). The RNA is stably bound to the nitrocellulose paper by this procedure, allowing removal of the hybridized probes and rehybridization of the RNA blots without loss of sensitivity. The use of nitrocellulose paper for the
analysis of RNA by blot hybridization has several advantages over the use of activated paper (diazobenzyloxymethyl-paper). The method is simple, inexpensive, reproducible, and sensitive. In addition, denaturation of DNA with glyoxal and dimethyl sulfoxide promotes transfer and retention of small DNAs (100 nucleotides and larger) to nitrocellulose paper. A related method is also described for dotting RNA and DNA directly onto nitrocellulose paper treated with a high concentration of salt; under these conditions denatured DNA of less than 200 nucleotides is retained and hybridizes efficiently. 3.GENOMIC SEQUENCING Unique DNA sequences can be determined directly from mouse genomic DNA. A denaturing gel separates by size mixtures of unlabeled DNA fragments from complete restriction and partial chemical cleavages of the entire genome. These lanes of DNA are transferred and UV-crosslinked to nylon membranes. Hybridization with a short 32P-labeled single-stranded probe produces the image of a DNA sequence "ladder" extending from the 3' or 5' end of one restriction site in the genome. Numerous different sequences can be obtained from a single membrane by reprobing. Each band in these sequences represents 3 fg of DNA complementary to the probe. Sequence data from mouse immunoglobulin heavy chain genes from several cell types are presented. The genomic sequencing procedures are applicable to the analysis of genetic polymorphisms, DNA methylation at deoxycytidines, and nucleic acid-protein interactions at single nucleotide resolution 4.SUPERCOIL SEQUENCING:A FAST AND SIMPLE METHOD FOR SEQUENCING PLASMID DNA A method for obtaining sequence information directly from plasmid DNA is presented. The procedure involves the rapid preparation of clean supercoiled plasmid DNA from small bacterial cultures, its complete denaturation by alkali, and sequence determination using oligodeoxyribonucleotide-primed enzymatic DNA synthesis in the presence of dideoxynucleoside triphosphates. The advantages of the method include speed, simplicity, avoidance of additional cloning steps into single-stranded phage M13 vectors, and hence applicability to sequencing large numbers of samples. 5. HIGH EFFICIENCY TRANSFORMATION OF INTACT YEAST CELLS USING SINGLE STRANDED NUCLEIC ACIDS AS A CARRIER A method, using LiAc to yield competent cells, is described that increased the efficiency of genetic transformation of intact cells of Saccharomyces cerevisiae to more than 1 X 10(5) transformants per microgram of vector DNA and to 1.5% transformants per viable cell. The use of single stranded, or heat denaturated double stranded, nucleic acids as carrier resulted in about a 100 fold higher frequency of transformation with plasmids containing the 2 microns origin of replication. Single stranded DNA seems to be responsible for the effect since M13 single stranded DNA, as well as RNA, was effective. Boiled carrier DNA did not yield any increased transformation efficiency using spheroplast formation to induce DNA uptake, indicating a difference in the mechanism of transformation with the two methods. 6.DIDEOXY SEQUENCING METHOD USING DENATURED PLASMID TEMPLATES The dideoxy sequencing method in which denatured plasmid DNA is used as a template was improved. The method is simple and rapid: the recombinant plasmid DNA is
extracted and purified by rapid alkaline lysis followed by ribonuclease treatment. The plasmid DNA is then immediately denatured with alkali and subjected to a sequencing reaction utilizing synthetic oligonucleotide primers. It takes only several hours from the start of the plasmid extraction to the end of the sequencing reaction. We examined each step of the procedure, and several points were found to be crucial for making the method reproducible and powerful: (i) the plasmid DNA should be free from RNA and open circular (or linear) DNA; (ii) a heptadecamer rather than a pentadecamer is recommended as a primer; and (iii) the sequencing reaction should be done at 37 degrees C or higher rather than at room temperature. The method enabled us to determine the sequence of more than a thousand nucleotides from a single template DNA. ADVANTAGES OF RENATURATION OF DNA 1.) Renaturation of DNA in the presence of ethidium bromide
The rate of renaturation of T2 DNA has been studied as a fuction of ethidium bound per nucleotide of denatured DNA. The Binding constants and number of binding sites for ethidium have been determined by spectral titration for denatured DNA at 55, 65, and 75C and for native DNA at 65C in 0.4M Na+. The rate of renaturation of T2 DNA was found to be independentof ethidium binding up to 0.03 moles per mole of nucleotide. Above 0.03 moles, the rate drops off precipitously approaching zero at 0.08 and 0.06 moles bound ethidium per nucleotide at 65C respectively. A study was also made of the use of bound ethidium fluorescence as a probe for monitoring DNA renaturation reactions. 2) One hundred-fold acceleration of DNA renaturation rates in solution Solvents which accelerate DNA renaturation rates have been investigated. Addition of NaCl or LiCl to DNA in 2.4M Et4NCl initially increases renaturation rates at 45C and then leads to a loss of second-order behavior. The greatest accelerations are seen with LiCl and dilute DNA. Volume exclusion by dextran sulfate is the most effective method of accelerating DNA renaturation with concentrated DNA. Addition of dextran sulfate beyond 10-12% in 2.4M Et4NCl fails to increase the acceleration beyond approximately 10-fold. Accelerations of 100-fold may be achieved with 3540% dextran sulfate in 1M NaCl at 70C. No other mixed solvent system was found to be more effective, although acceleration may be achieved in solvents containing formamide or other denaturants. The acceleration in 2M NaCl occurs without loss of the normal concentration and temperature dependence of DNA renaturation and is also independent of dextran sulfate concentration if sufficient dextran sulfate is used. Dextran sulfate may be selectively precipitated by use of 1M CsCl. 3) Renaturation and Hybridization Studies of Mitochondrial DNA The products of the renaturation reaction of mitochondrial DNA from oocytes of Xenopus laevis have been studied by electron microscopy and CsCl equilibrium density gradient centrifugation. The reaction leads to the formation of intermediates containing single-stranded and double-stranded regions. Further reactions of these intermediates result in large complexes of interlinking doublestranded filaments. The formation of circular molecules of the same length as native circles of mitochondrial DNA was also observed. The formation of common high molecular weight complexes during joint reannealing of two DNA's with complementary sequences was used as a method to detect sequence homology in
different DNA samples. Although this method does not produce quantitative data it offers several advantages in the present study. No homologies could be detected between the nuclear DNA and the mitochondrial DNA of X. laevis or of Rana pipiens. In interspecies comparisons homologies were found between the nuclear DNA's of X. laevis and the mouse and between the mitochondrial DNA's of X. laevis and the chick, but none between the mitochondrial DNA's of X. laevis and yeast. These results are interpreted as indicating the continuity of mitochondrial DNA during evolution.
Chapter-23
Safety and regulatory aspects of enzyme use
Introduction Only very few enzymes present hazards, because of their catalytic activity, to those handling them in normal circumstances but there are several areas of potential hazard arising from their chemical nature and source. These are allergenicity, activity-related toxicity, residual microbiological activity, and chemical toxicity. All enzymes, being proteins, are potential allergens and have especially potent effects if inhaled as a dust. Once an individual has developed an immune response as a result of inhalation or skin contact with the enzyme, re-exposure produces increasingly severe responses becoming dangerous or even fatal. Because of this, dry enzyme preparations have been replaced to a large extent by liquid preparations, sometimes deliberately made viscous to lower the likelihood of aerosol formation during handling. Where dry preparations must be used, as in the formulation of many enzyme detergents, allergenic responses by factory workers are a very significant problem particularly when fine-dusting powders are employed. Workers in such environments are usually screened for allergies and respiratory problems. The problem has been largely overcome by encapsulating and granulating dry enzyme preparations, a procedure that has been applied most successfully to the proteases and other enzymes used in detergents. Enzyme producers and users recognise that allergenicity will always be a potential problem and provide safety information concerning the handling of enzyme preparations. They stress that dust
in the air should be avoided so weighing and manipulation of dry powders should be carried out in closed systems. Any spilt enzyme powder should be removed immediately, after first moistening it with water. Any waste enzyme powder should be dissolved in water before disposal into the sewage system. Enzyme on the skin or inhaled should be washed with plenty of water. Liquid preparations are inherently safer but it is important that any spilt enzyme is not allowed to dry as dust formation can then occur. The formation of aerosols (e.g. by poor operating procedures in centrifugation) must be avoided as these are at least as harmful as powders. Activity-related toxicity is much rarer but it must be remembered that proteases are potentially dangerous, particularly in concentrated forms and especially if inhaled. No enzyme has been found to be toxic, mutagenic or carcinogenic by itself as might be expected from its proteinaceous structure. However, enzyme preparations cannot be regarded as completely safe as such dangerous materials may be present as contaminants, derived from the enzyme source or produced during its processing or storage. The organisms used in the production of enzymes may themselves be sources of hazardous materials and have been the chief focus of attention by the regulatory authorities. In the USA, enzymes must be Generally Regarded As Safe (GRAS) by the FDA (Food and Drug Administration) in order to be used as a food ingredient. Such enzymes include -amylase,-amylase, bromelain, catalase, cellulase, ficin, galactosidase, glucoamylase, glucose isomerase, glucose oxidase, invertase, lactase, lipase, papain, pectinase, pepsin, rennet and trypsin. In the UK, the Food Additives and Contaminants Committee (FACC) of the Ministry of Agriculture, Fisheries and Food classified enzymes into five classes on the basis of their safety for presence in the foods and use in their manufacture. Group A. Substances that the available evidence suggests are acceptable for use in food. Group B. Substances that on the available evidence may be regarded as provisionally acceptable for use in food but about which further information must be made available within a specified time for review. Group C. Substances for which the available evidence suggests toxicity and which ought not to be permitted for use in food until adequate evidence of their safety has been provided to establish their acceptability. Group D. Substances for which the available information indicates definite or probable toxicity and which ought not to be permitted for use in food. Group E. Substances for which inadequate or no toxicological data are available and for which it is not possible to express an opinion as to their acceptability for use in food. This classification takes into account the potential chemical toxicity from microbial secondary metabolites such as mycotoxins and aflotoxins. The growing body of knowledge on the long-term effects of exposure to these toxins is one of the major reasons for the tightening of legislative controls. The enzymes that fall into group A are exclusively plant and animal enzymes such as papain, catalase, lipase, rennet and various other proteases. Group B contains a very wide range of enzymes from microbial sources, many of which have been used in food or food processing for many hundreds of years. The Association of Microbial Food Enzyme Producers (AMFEP) has suggested subdivisions of the FACC's group B into: Class ain8 microorganisms that have traditionally been used in food or in food processing, including Bacillus subtilis, Aspergillus niger, Aspergillus oryzae, Rhizopus oryzae, Saccharomyces cerevisiae, Kluyveromyces fragilis, Kluyveromyces lactis and Mucor javanicus. Class bin8 microorganisms that are accepted as harmless contaminants present in food, including Bacillus stearothermophilus, Bacillus licheniformis, Bacillus coagulans, and Klebsiella in8aerogenes. Class cin8 microorganisms that are not included in Classes b and c, including Mucor miehei, Streptomyces albus, Trichoderma reesei, Actinoplanes missouriensis,
and Penicillium emersonii. It was proposed that Class a should not be subjected to testing and that Classes b and c should be subjected to the following tests: acute oral toxicity in mice and rats, subacute oral toxicity for 4 weeks in rats, oral toxicity for 3 months in rats, and in vitro mutagenicity. In addition Class c should be tested for microorganism pathogenicity and, under exceptional circumstances, in vivo mutagenicity, teratogenicity, and carcinogenicity. The cost of the various tests needed to satisfy the legal requirements are very significant and must be considered during the determination of process costs. Plainly the introduction of an enzyme from a totally new source will be a very expensive matter. It may prove more satisfactory to clone such an enzyme into one of AMFEP's Class a organisms but this will first require new legislation to regulate the use of cloned microbes in foodstuffs. Some of the safety problems associated with the use of free enzymes may be overcome by using immobilised enzymes . This is an extremely safe technique, so long as the materials used are acceptable and neither they, nor the immobilised enzymes, leak into the product stream.
ENZYMOLOGY AND ENZYME TECHNOLOGY Course Code: BTBBT 30602
Course Objective: The course aims to provide an understanding of the principles and application of proteins, secondary metabolites and enzyme biochemistry in therapeutic applications and clinical diagnosis. The theoretical understanding of biochemical systems would certainly help to interpret the results of laboratory experiments. Course Contents: Module I Enzymes: Introduction and scope, Nomenclature, Mechanism of Catalysis. Module II Specificity of enzyme action, monomeric and oligomeric enzymes, Enzyme inhibition. Module III Enzyme Kinetics: Single substrate steady state kinetics; Michaelis Menten equation, Linear plots, King-Altmans method; Inhibitors and activators; Multisubstrate systems; ping-pong mechanism, Alberty equation, Sigmoidal kinetics
and Allosteric enzymes Module IV Extraction & purification of enzymes. Module V Immobilization of Enzymes; Advantages, Carriers, adsorption, covalent coupling, cross-linking and entrapment methods, Micro-environmental effects. Module VI Biotechnological applications of enzymes: Large scale production and purification of enzymes, enzyme utilization in industry, enzymes and recombinant DNA technology Examination Scheme: Component Codes Weight age (%) H/Q 10 S 10 CT2 20 EE 60
Text & References: Text Biotechnological Innovations in Chemical Synthesis, R.C.B. Currell, V.D. Mieras, Biotol Partners Staff, Butterworth Heinemann. Enzyme Technology, M.F. Chaplin and C. Bucke, Cambridge University Press. Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis, R.A. Copeland, John Wiley and Sons Inc. References Enzymes Biochemistry, Biotechnology, Clinical Chemistry, Trevor Palner Dr. S. M. Bhatt smbhatt@amity.edu , smbhatt_bhu@rediffmail.com phone 9313993840 Amity University Uttar Pradesh Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems, I.H. Segel, Wiley-Interscience Industrial Enzymes & their applications, H. Uhlig, John Wiley and Sons Inc. ENZYMOLOGY AND ENZYME TECHNOLOGY LAB Course Code: BTBPL 30622
Course Objective: The laboratory will help the students to isolate enzymes from different sources, enzyme assays and studying their kinetic parameters which have immense importance in industrial processes. Course Contents: Module I Isolation of enzymes from plant and microbial sources. Module II Enzyme assay; activity and specific activity determination of amylase, nitrate reductase, cellulase, protease Module III Purification of Enzyme by ammonium sulphate fractionation
Module IV Enzyme Kinetics: Effect of varying substrate concentration on enzyme activity, determination of Michaelis-Menten constant (Km) and Maximum Velocity (Vmax.) using Lineweaver-Burk plot. Module V Effect of Temperature and pH on enzyme activity Module VI Enzyme immobilization Examination Scheme: Major Experiments: Minor Experiments: Spotting: Viva: Records: Total: 40 20 10 20 10 100
Note: Minor variation could be there depending on the examiner. Text: Practical Biochemistry, Sawhney and Singh
Chapter- 1
Enzymes an introduction 9
Definition Introduction Historical aspects in enzyme discovery Classification of Enzymes. application of enzymes Chapter-2 Types of Enzymes 16 A. Simple enzymes B. Complex enzymes Complex enzymes Role of Coenzymes A. Catalysts B. Biocatalyst or Enzymes How enzymes speed up the reaction without taking part Unit of activity Chapter 3 Enzyme Specificity 20 ENZYME MECHANISM MODELS A. Lock and Key Model for Enzyme Activity B. Induced Fit (Hand and Glove) Model of Enzyme Activity Mechanisms of Enzyme Catalysis Chapter 4 Enzyme-Substrate Interactions 24 a. Catalysis by Bond Strain b. Catalysis by Proximity and Orientation c. Catalysis Involving Proton Donors (Acids) and Acceptors (Bases) Concerted acid-base catalysis d. Covalent Catalysis:
Covalent Intermediates e) Metal Ion Catalysis Catalytic Power a) Proximity b) Orientation f. Catalysis in organic solvents Some advantages to using enzymes in non-aqueous solvents include (Dordick, 1989): Importance and relevance of whole cell enzymes in biotechnology There may be some drawbacks to using whole cells in catalysis. These include Abzymes Exercises Chapter-5 ENZYME KINETICS & INHIBITION 46 Chemical Reactions and Rates Chemical Reaction Order Michaelis-Menton constant a. Pre-steady state:-b. Steady state:-Derivation: Catalytic efficiency and Turnover number. Chapter-6 Factor affecting the rate of catalysis 58 Substrate concentration Effect of temperature and pressure Effect of pH on Enzyme Chapter-7 Enzyme inhibition 68 A. Competitive inhibition B. Uncompetitive inhibition C. Noncompetitive inhibition Determination of Vmax and Km Chapter-8 Multisubstrate enzymes 83 Enzyme Catalysis Mechanisms of Chemical Catalysis one-substrate enzymes: Linear plots for enzyme kinetic studies FYI - The Eadie-Hofstee Plot ENZYME KINETICS AND INHIBITION What's exciting about enzyme inhibition? Multi-substrate Enzymes Chapter-9 ISOLATION of ENZYME 102 INTRODUCTION strategies for enzyme purification. Method of separation. Test of purity is done by following method Source of enzyme Media for enzyme production Cell can be break down byfollowing methods A. Cell breakage by osmotic shock B. Use of enzymic lytic methods c. Ultrasonic cell disruption d. Cell lysis by High pressure homogenisers Method of separation of enzyme from lysed cell; CentrifugationB. ULTRACENTRIFUGATION A. Differential centrifugation Filtration of enzyme after separation Separation of enzyme in Aqueous biphasic systems
Preparation of enzymes from clarified solution; Ultrafiltration Chapter-10 Concentration and purification of enzyme 123 Concentration by precipitation A. Nucleic acid removal B. Heat treatment 1. Enzyme purification by Chromatography 2 Affinity chromatography .3 Hydrophobic interaction chromatography (HIC) or affinity elution CHOICE OF MATRIX Electrophoresis 2.11 Maintaining Enzyme Activity Chapter-11 Techniques used in Enzyme characterization 132 HOW TO KNOW THE PROPERTY OF ENZYME Introduction A. Chromatography Principles of chromatography Adsorption Chromatography COLUMN CHROMATOGRAPHY History Principle Mechanism of separation by size in gel permeation chromatography ADVANTAGE OF GEL CHROMATOGRAPHY Application of GPC or gel permeation chromatography PAPER CHROMATOGRAPHY Ion exchange chromatography There are two type of ion exchanger Choosing an Ion Exchanger Gas / Liquid Chromatography Reversed Phase Chromatography HIGH PRESSURE LIQUID CHROMATOGRAPHY [HPLC] Partition Chromatography Affinity chromatography Principle METHOD OF LIGAND IMMOBILIZATION 1. CNBR activated agarose 2. 6-AMINO-HEXENOIC ACID & 1,6 DIAMINO HEXANE. 3. Carbomyl diimidazole activated agarose. Applications Thin layer chromatography Principle ADVANTAGE OF TLC OVER OTHER CHROMATOGRAPHY TECHNIQUE HPTLC and HPPLC Selection of chromatographic system Electrophoretic technique a. Agarose Gel Electrophoresis b. Polyacrylamide Gel Electrophoresis c. SDS-PAGE f. Iso-electric focusing: IEF gel: g. Chromatofocussing 2D-PAGE DETECTION, ESTIMATION AND RECOVERY OF PROTEIN GELS. i. AGAROSE GEL ELECTROPHORESIS OF DNA Preparation of the gel Polyacrylamide gel Chapter -12 Enzyme Immobilization 160
127
Introduction Aim of Enzyme Immobilization [a] Physical properties [b] Chemical properties [c] Stability [d] Resistance [e] Safety [f] Economic Limitations of Immobilized Enzyme Advantages of Immobilizations Methods of immobilizations Carrier matrices Adsorption of enzymes Some disadvantages of adsorption include Covalent coupling Functional groups that affects the covalent coupling A. cyanogen bromide (b) Ethyl chloroformate (c) Carbodiimide (d) Glutaraldehyde (e) 3-aminopropyltriethoxysilane Entrapment and Encapsulation Entrapment of enzymes Membrane confinement Encapsulation of Enzymes Crosslinking Chapter-13 Introduction to Industrial biotechnology 171 Application of Immobilised-Enzyme Processes Introduction 172 1-High-fructose corn syrups (HFCS) 2-GLUCOSE ISOMERASE a Treatment with activated carbon. 3-Use of immobilised raffinase 4-Use of immobilised Invertase 5-Production of amino acids 6- Use of immobilised lactase .7 Production of antibiotics Preparation of acrylamide Chapter-14 Application of Enzyme in clinics 233 Introduction Determination of enzyme activities 1- Clinical Enzymology of liver disease. 2- In Heart disease -amylase Creatine Kinase and fructose bisphosphate aldolase. Alkaline phosphates Acid phosphatase. In Treatment of cancer Enzyme deficiencies Enzyme inhibitors and drug design 183 Use of enzyme in Dignosis Blood glucose Disadvantages of Using Enzyme as Therapeutic Agents Thermozymes The large-scale use of enzymes in solution The use of enzymes in detergents Chapter-15Application of Enzyme in Industries
different sector of industry where enzymes are used Food Textile and other indsutries Chapter-16 Enzyme Reactors all type of reactors detailed dscription kinetics involved in reactors Chapter-17 Protein Engineering 215 Artificial enzymes Chapter-18 Biosensors and Immunosensors 225 Chapter-19 Enzyme and protein STABILITY 246 I Definition of Stability II The Unfolded State III Major Factors Affecting Protein Stability The Hydrophobic Effect Hydrogen Bonds Conformational Entropy of Unfolding IV Other Factors Affecting Protein Stability Salt Bridges Aromatic-Aromatic Interactions Metal Binding Disulphide Bonds V Chemical Degradation VI Conclusions Chapter -20 Study of drug and molecule binding stratgies 260 TYPES OF DNA BINDING DRUGS NON COVALENT INTERACTIONS Minor Groove binders INTERCALATORS COVALENT INTERACTIONS DNA-Drug Interaction : Chapter-21 Technique to study protein 284 Nuclear Magnetic Resonance(NMR)Spectroscopy Determination of membrane protein structure by rotational resonance NMR: bacteriorhodopsin. Structure of bacteriorhodopsin: Determination of membrane protein structure by rotational resonance NMR: bacteriorhodopsin Chapter-22 Enzymology of DNA folding and unfolding 290 WHAT IS DNA Major and minor grooves Replication Enzyme involved in DNA folding and unfolding Invitro-DNA folding and unfolding Folding unfolding kinetics Chapter-23 Safety and regulatory aspects of enzyme use 305
Module I Enzymes: Introduction and scope, Nomenclature, Mechanism of Catalysis. Chapter- 1 Enzymes an introduction Definition Calorimetry: Measurement of heat released or taken up in a chemical reaction or physical process to derive thermodynamic data (e.g., dissociation constant, freeenergy change, enthalpy change, entropy change) for molecular interactions. Two kinds of calorimetry important to biomolecular studies are isothermal titration calorimetry (ITC), which can be used to detect the number of binding sites on an enzyme; and differential scanning calorimetry (DSC), which monitors. Enzyme: Protein that acts as a catalyst, speeding the rate of a biochemical reaction but not altering its direction or nature. Electrophoresis: Method of separating large molecules (such as DNA fragments or proteins) in a sample. An electric current is passed through a medium containing the sample; each molecule travels through the medium at a different rate, depending on its electrical charge, shape and size. Agarose and acrylamide gels are commonly used media for electrophoresis of proteins and nucleic acids. In onedimensional (1D) gel electrophoresis, proteins and nucleic acids are separated in one direction on a gel, primarily by size. Two-dimensional (2D) gel electrophoresis is used in proteome analyses to separate complex protein mixtures using two separation planes (e.g., vertically down a gel by net charge and horizontally by molecular mass). Each unique protein mixture produces a characteristic pattern or fingerprint of protein separation on a 2D gel. Flow Cytometry: Analysis of biological material by detection of light-absorbing or fluorescing properties of cells or subcellular components (e.g., chromosomes) passing in a narrow stream through a laser beam. An absorbance or fluorescence profile of the sample is produced. Automated sorting devices, used to fractionate samples, separate successive droplets of the analyzed stream into different fractions depending on the fluorescence emitted by each droplet. Kinetics: Field of study that deals with determining the rates of biological, chemical, and physical processes (e.g., how quickly reactants are converted into products) under various conditions. Metalloprotein: Protein that incorporates one or more metals into its molecular structure by binding individual metal ions [e.g., iron (Fe2+ or Fe3+), zinc (Zn2+), or magnesium (Mg2+)] or nonprotein organic compounds containing metals. Metalloprotein are important components of electron transport chains. Steady State: Growth state in which the concentration of bacterial cells is in equilibrium with the concentration of nutrients or substrates (i.e., the concentrations remain constant over time). X-ray crystallography: Technique used to obtain structural information for a substance (e.g., protein, molecular complex) that has been crystallized. A beam of X rays is focused on the crystals, and the scattering pattern of the X rays is used to create 3D representations of the crystal with atomic resolution.
Introduction Enzymes are the wonderful creations of the biological system. Various biological reactions go on smoothly without single mistakes. Enzymes have specificity not only toward the substrate but also recovered in the end of the reactions. They are also regulated very precisely. Normal monomeric enzyme like trypsin and pepsin are regulated by removing or adding a small piece of peptide that make them inactive or active while the complex enzymes are regulated by end product , or by allosteric mechanism. Homones in several cases also play important role in regulations like in formation of lactose by progestron and prolactin before and after the birth of baby. The system (complex enzymes are termed as system) specifically and precisely binds to the substrate and convert them into the required product that sometimes becomes the substrate for next reaction for next enzyme to act on. Total activity of enzyme depends on several factors like acid base, structural orientations and strain, electrostatic interactions (by metal ions) and formation of covalent bond. In several cases certain organic molecule or metals termed as co-enzyme or cofactor that either or electron or proton acceptor or by electrostatic mechanism are needed to convert a substrate to product. Actually enzymes activity depends on the precise function of a small part called as the active site that is a small pocket like structure that contains various helper amino acid residues that by electron or proton transport make bond of the substrate molecule weaker and rearrange them into a stable product at normal temperature and pressure. Any deficiency in the formation of these pockets like structure may render the whole enzyme to become inactive that leads to certain deficiency diseases. Modern techniques like MOLDI-TOF, x-ray crystallography along with sequencer helped a lot to scientist in deciphering the mechanism of enzyme activity. Knowledge of enzyme activity and its detailed mechanism made it possible to design artificial enzyme in the lab to cop up with many enzyme related metabolic problems or complex viral enzyme like AIDS. Historical aspects in enzyme discovery 1860 Bertholate disrupted yeast cell and isolated extract that catalyzed conversion of sucrose to glucose and fructose. 1878 Word enzyme first used by kuhne. 1894 Fisher proposed lock and key hypothesis to explain the mechanism of Enzyme operating between substrate and enzyme. 1897 Buchner isolated yeast extract used for formation of ethanol. 1920 Sumner crystallized Urease first time that hydrolyze the urea into CO2 and NH3. 1958 and 1960 1961 1965 1986 Koshland proposed induced fit model to account for enzyme catalytic power specificity. Ribonuclease amino acid was sequenced. Ribonuclease was artificially synthesized from amino acid precursor. three dimensional structure of Lysozyme was deduced. RNase was found to work as catalyst by Cech. They were called as ribozymes. e.g. Ribonuclease P
Classification of Enzymes. The I.U.B. system also specifies a textual name for each enzyme. The enzyme's name is comprised of the names of the substrate(s), the product(s) and the enzyme's functional class. Because many enzymes, such as alcohol dehydrogenase, are widely known in the scientific community by their common names, the change to I.U.B.approved nomenclature has been slow. In everyday usage, most enzymes are still called by their common name. In 1955 International Union of Biochemistry classified the enzyme on the basis of kind of reaction they performed, substrate they act, position of group or functional group and number of amino acid. Therefore they divided the enzyme into six major classes. Suffix added in the enzyme is ase to the name of substrate or to word. Urea hydrolysis is done by the Urease. There are major 6 classes and each with the subclass. Each enzyme has assigned 4 digit classification number and a systematic name. For e.g. EC 2.7.1.1 means first digit indicates the class transferases, and the second digit denotes the subclass, third and four digits is the group transfer or accepted as shown in figure. Classes of enzyme: there are six classes of enzyme according to reaction type they performed 1. Oxidoreductases- These are enzymes that catalyze oxidations or reductions. Oxidation means transfer of oxygen atom and reduction means transfer of H atom. Enzymes such as dehyrogenases, oxidases, and peroxidases. Its second digit has been denoted in the table. Its third digit refers to the hydrogen or electron acceptor. For example 1-for NAD+ or NADP+ 2. For Fe+3 3. For oxygen. For clear understanding we can take example of S-latate NAD+ oxidoreductase (E.C. 1.1.1.27). Here S lactate is the substrate and NAD+ is electron acceptor/ hydrogen acceptor and the reaction is of type oxidation reaction. In EC number 1 digit denotes type of reaction i.e. oxidoreductase 2nd digit is the functional group of lactate which is alcohol therefore it is denoted by subclass 1, 3rd digit denotes NAD as electron acceptor NAD+, while 4th digit is the actual substrate. Lactate dehydrogenase CH3CH(OH)COO- + NAD+ ---------- CH3C(O)COO- + NADH+ + H+ 2. Transferases- These enzymes catalyze the transfer of a group from one molecule to another. Examples such as Phosphatases, transaminases, and transmethylases. Its second digit has been provided in the table. Its third digit denotes the group transferred. Thus 1 denotes transfer of CH3 group while 2 denotes the transfer of CH2OH and 3 denotes the carboxyl or carbamoyl transferases. Phosphotransferases are given trivial name as kinase for example hexokinases (E.C. 2.7.1.1 or D-hexose-6-phosphotransferase, 7 for phosphate group, 1 for single carbon unit) that catalyses the transfer of phosphate from ATP to D hexose resulting the D hexose-6-phosphate. D-hexose-6-phosphotransferase (hexokinase) D hexose + ATP-----------------------------------phosphate + ADP D hexose-6-
3. Hydrolases-These enzymes catalyze hydrolysis reactions. Examples are the digestive enzymes such as sucrase, amylase, maltase, and lactase. They are classified according to the type of bond hydrolysed 1 for ester 2 for glycosidic bond, 3 for ether bond, 4 for peptide bond etc. third digit denotes the type of bond hydrolysed like 1 for hydrolysis of ester bond, 2 for thiol ester bond, 3 for monoester bond, 4 for phosphoric diester hydrolases. For example alkaline phophatases is a hydrolase that hydrolyse variety of substrate at alkaline pH.
4. Lyases- These enzymes catalyze the removal of groups and create double bond in non-aqueous media. An example would be the decarboxylases. Its second digit denotes the bond broken and third digit denotes type of group removed like 1 for carboxyl group and 2 for aldehyde group and 3 for ketoacid group. L histidine carboxy lyase histidine -----------------------------------histamine + CO2 5. Isomerases- Enzymes that catalyze the isomerization of molecules means L isomer will be D isomer after catalysis and cis isomer will be trans isomer after the catalysis for Examples L alanine will be converted into the D alanine by alanine racemases. 2nd digit refers to the type of reaction like 1 for recemization or epimerization (if inversion occurs at assymitric carbon having four different attached group. 6. Ligases- These are also called synthetases which are enzymes that catalyze condensation reactions where smaller molecules are connected with the resulting removal of a water molecule. This is accompanied with the formation of a high energy Phosphate link that stores energy. An example would be the amino acid RNA Ligases. Subclass Name EC 1 Oxidoreductases EC 1.1 Acting on the CH-OH group of donors EC 1.2 Acting on the aldehyde or oxo group of donors EC 1.3 Acting on the CH-CH group of donors EC 1.4 Acting on the CH-NH2 group of donors EC 1.5 Acting on the CH-NH group of donors EC 1.6 Acting on NADH or NADPH EC 1.7 Acting on other nitrogenous compounds as donors EC 1.8 Acting on a sulfur group of donors EC 1.9 Acting on a heme group of donors EC 1.10 Acting on diphenols and related substances as donors EC 1.11 Acting on a peroxide as acceptor EC 1.12 Acting on hydrogen as donor EC 1.13 Acting on single donors with incorporation of molecular oxygen (oxygenases) EC 1.14 Acting on paired donors, with incorporation or reduction of molecular oxygen EC 1.15 Acting on superoxide radicals as acceptor EC 1.16 Oxidising metal ions EC 1.17 Acting on CH or CH2 groups EC 1.18 Acting on iron-sulfur proteins as donors EC 1.19 Acting on reduced flavodoxin as donor EC 1.20 Acting on phosphorus or arsenic in donors EC 1.21 Acting on X-H and Y-H to form an X-Y bond EC 1.97 Other oxidoreductases EC 2 Transferases EC 2.1 Transferring one-carbon groups EC 2.2 Transferring aldehyde or ketonic groups EC 2.3 Acyltransferases EC 2.4 Glycosyltransferases EC 2.5 Transferring alkyl or aryl groups, other than methyl groups EC 2.6 Transferring nitrogenous groups EC 2.7 Transferring phosphorus-containing groups EC 2.8 Transferring sulfur-containing groups EC 2.9 Transferring selenium-containing groups
EC 3 Hydrolases EC 3.1 EC 3.2 EC 3.3 EC 3.4 EC 3.5 EC 3.6 EC 3.7 EC 3.8 EC 3.9 EC 3.10 EC 3.11 EC 3.12 EC 3.13 EC 4 Lyases EC 4.1 EC 4.2 EC 4.3 EC 4.4 EC 4.5 EC 4.6 EC 4.99 EC 5 Isomerases EC 5.1 EC 5.2 EC 5.3 EC 5.4 EC 5.5 EC 5.99 EC 6 Ligases EC 6.1 EC 6.2 EC 6.3 EC 6.4 EC 6.5 EC 6.6 SN 1 2 3 4 5 6
Acting on ester bonds Glycosylases Acting on ether bonds Acting on peptide bonds (peptidases) Acting on carbon-nitrogen bonds, other than peptide bonds Acting on acid anhydrides Acting on carbon-carbon bonds Acting on halide bonds Acting on phosphorus-nitrogen bonds Acting on sulfur-nitrogen bonds Acting on carbon-phosphorus bonds Acting on sulfur-sulfur bonds Acting on carbon-sulfur bonds Carbon-carbon lyases Carbon-oxygen lyases Carbon-nitrogen lyases Carbon-sulfur lyases Carbon-halide lyases Phosphorus-oxygen lyases Other lyases Racemases and epimerases cis-trans-Isomerases Intramolecular isomerases Intramolecular transferases (mutases) Intramolecular lyases Other isomerases Forming Forming Forming Forming Forming Forming carbonoxygen bonds carbonsulfur bonds carbonnitrogen bonds carboncarbon bonds phosphoric ester bonds nitrogenmetal bonds
Class Reaction catalyzed.
Oxidoreductases Transferases
Hydrolases Lyases Isomerases Ligase Electron transfer Group transfer Hydrolysis Addition of group to double bonds Transfer of group to yield isomeric forms. Formation of C-C,C-S,C-O,C-N. Some metalloenzymes and their cofactor Enzyme Cofactor Cytochrome oxidase Catalase Peroxidase. Cytochrome oxidase Carbonic anhydrase Alcohol dehydrogenase Hexokinase Glucose -6-phosphate Pyruvate kinase. Arginase Ribonucleotide reductase Pyruvate kinase. Urease Dinitrogenase Glutathione peroxidase Fe+2 or Fe+3
Cu+2 Zn+2 Mg+2
Mn+2 K+ Ni+2 Mo Se In addition to the classification name most enzymes have group names. Hydrolases-Enzymes that catalyze hydrolysis reactions. hydrogenases- Enzymes that catalyze the addition of Hydrogen atoms Oxidases- Enzymes that catalyze oxidations In addition each enzyme has a specific name which usually consists of part of the name of the substrate molecule. Examples are sucrase, Maltase, Lactase, galactosidase, and RNAase. Other enzymes have retained common names attributed to them. Enzymes such as, Pepsin, Chymotrypsin, Trypsin, Catalase, Alpha Amylase, Lysozyme, and others. Chapter-2 Types of Enzymes
A. Simple enzymes They are composed solely of protein, single or multiple subunits. If they consist of single polypeptide unit they are termed as monomeric enzyme ( sometime like in chymotrypsinogen they are of three polypeptide and are inctive zymogen form and after cleavage from pie to delta and finally to alpha trypsinogen they become active and consist of two polypepide and still they are termed as monomeic enzyme since they are read in continous sequences). Monomeric enzymes are of few 200-300 amino acid and molecular weight ranges from 15,000 kd to 35,000 kd (kilodalton) like trypsin , elastases, that are termed as serine proteases because of an active serine residue. Serine residue become active due to relay mechanism of electron transfer (adjacent amino acid can transfer electron in series of pathway and make the serine active sufficient to active at electron deficient or carbocation center of substrate making breaking of bond possible. Thus a slight decrease in entropy make free energy negative that is essential for stabilization of intermediate transition state). Other enzyme consisting of two or more polypeptide are termed as oligomeric enzyme. Like alcohol dehydrogenase (contains two polypeptide and Zn+2 and act by electrostatic meechansim) and lactate dehydrogenase that requires cofactor NAD+ or NADP+ for its activity and occurs in multiple unit like H4, H3M, H2M2, HM3, and M4. Thus they contain four subunit and substrate can bind to each of four subunit and they are termed as isozyme. Actually Ogstien considered three sites on enzyme one or two binding site along with one catalytic subunit. Lactate is formed from pyruvate in aneorbic condition and thus pyruvate accumulation are avoided. Lactate arev then transported to other organs. B. Complex enzymes Complex enzymes They involve protein plus small organic molecule(s) or metal. Entire complex called holoenzyme. Protein part called apoenzyme. Non-protein part called coenzyme or prosthetic Group. They form the integral part of the enzyme and remain adjacent to the catalytic center to provide coordination for interacting with substrate functional group. Therefore multiple substrates can interact with
enzyme and react with them. The detailed mechanism will be discussed in next topic under the head of enzyme specificities. These rules give each enzyme a unique number. Enzymes are also classified on the basis of their composition. Enzymes composed completely of protein are known as simple enzymes in contrast to complex enzymes, which are composed of protein plus a relatively small organic molecule. Complex enzymes are also known as holoenzymes. In this terminology the protein component is known as the apoenzyme, while the non-protein component is known as the coenzyme or prosthetic group where prosthetic group describes a complex in which the small organic molecule is bound to the apoenzyme by covalent bonds; when the binding between the apoenzyme and non-protein components is non-covalent, the small organic molecule is called a coenzyme. Many prosthetic groups and coenzymes are water-soluble derivatives of vitamins. It should be noted that the main clinical symptoms of dietary vitamin insufficiency generally arise from the malfunction of enzymes, which lack sufficient cofactors derived from vitamins to maintain homeostasis. The non-protein component of an enzyme may be as simple as a metal ion or as complex as a small non-protein organic molecule. Enzymes that require a metal in their composition are known as metalloenzymes if they bind and retain their metal atom(s) under all conditions that is with very high affinity. Those have a lower affinity for metal ion, but still require the metal ion for activity, are known as metal-activated enzymes. Role of Coenzymes The functional role of coenzymes is to act as transporters of chemical groups from one reactant to another. The chemical groups carried can be as simple as the hydride ion (H+ + 2e-) carried by NAD or the mole of hydrogen carried by FAD; or they can be even more complex than the amine (-NH2) carried by pyridoxal phosphate. Since coenzymes are chemically changed as a consequence of enzyme action, it is often useful to consider coenzymes to be a special class of substrates, or second substrates, which are common to many different holoenzymes. In all cases, the coenzymes donate the carried chemical grouping to an acceptor molecule and are thus regenerated to their original form. This regeneration of coenzyme and holoenzyme fulfills the definition of an enzyme as a chemical catalyst, since (unlike the usual substrates, which are used up during the course of a reaction) coenzymes are generally regenerated. A. Catalysts Catalysts are substances that increase product formation by lowering the energy barrier (activation energy) for the product to form and increase the favorable orientation of colliding reactant molecules for product formation to be successful. There are two types of catalysts: 1. Heterogeneous Catalysts 2. Homogeneous Catalysts Heterogeneous catalysts are those that provide a surface for the reaction to proceed upon. The catalyst and the reactant molecules are not in the same phase. This is sometimes referred to as surface catalysts. Certain transition state metals like Palladium, Platinum, Nickel, and Iron serve as industrial catalysts that catalyze a wide variety of reactions such as Hydrogenation. Homogeneous catalysts are catalysts that exist in the same phase as the reactant molecules usually in a solution. Acids and Bases in solution serve as catalysts in a wide variety of Organic reactions. Most industrial catalysts are responsible for more than one catalysis among reactants and are considered relatively non-specific in what they catalyze. Enzymes are very different. All current research suggests that enzymes are extremely specific in what a given enzyme catalyzes. Indeed, most enzyme molecules catalyze only one specific reaction but it does so in a
phenomenally efficient manner. One enzyme molecule might be responsible for converting thousands of reactant molecules called substrate molecules into product. B. Biocatalyst or Enzymes Enzymes are the protenaceous catalyst of the biosphere. Enzymes increase the rate of reactions without involving themselves being altered in the process of substrate conversion to product They mediate the formation of several biomolecules like Polynucleotide (DNA, RNA) protein enzyme, starch in plants, ATP etc. The Biological Catalysts are efficient in lowering down the activation energy of the reaction and thus they occur at very low temperature and pressure. All the chemical reactions inside the cell take place with the help of enzyme. Enzyme mediates rearrangement of the bond in substrate molecule by breaking of one covalent bond and formation of other covalent bond. The bond of the substrate molecule comes under strain due to presence of several functional group of different type over amino acid residue present inside the active site of the enzyme. Enzymes are biological catalysts responsible for supporting almost all of the chemical reactions that maintain animal homeostasis. Because of their role in maintaining life processes, the assay and pharmacological regulation of enzymes have become key elements in clinical diagnosis and therapeutics. The macromolecular components of almost all enzymes are composed of protein, except for a class of RNA modifying catalysts known as ribozymes. Ribozymes are molecules of ribonucleic acid that catalyze reactions on the Phosphodiester bond of other RNAs. Enzymes are found in all tissues and fluids of the body. Intracellular enzymes (enzyme located inside the cell and can be isolated after breaking the wall of the cell) catalyze the reactions of metabolic pathways. Plasma membrane enzymes (membrane bound enzyme) regulate catalysis within cells in response to extracellular signals (signal may be any chemical or any substrate molecule), and enzymes of the circulatory system are responsible for regulating the clotting of blood. Almost every significant life process is dependent on enzyme activity. Enzyme may occur anywhere inside the cell, like cell cytoplasm, vacuoles, and cell organelle. They are found in all the living system except the viruses. Enzymes are active at certain optimum temperature, pH. Enzymes can be active at higher temperature and pH if isolated from the extremophiles bacteria living in the harsh condition. One such enzyme is utilized in the PCR polymerase chain reaction, the taq polymerase isolated from the thermus aquaticus. Enzymes mostly mediate formation of specific isomer either L (+) or D (-). They are specific towards the substrate and remains in inactive form inside the cell. Their isolation involves the breaking of cell membrane, and their supernatant is utilized for further purification and isolation by NH4 SO4. In recent years Enzymes are in more demand due to their application in various biotech and pharmacy industry. In old days renine (new name chymosine) were utilized for the formation of cheese from milk. Most of the biological molecules are stable in the neutral pH, mild temp, and aqueous environment, found inside the cell. An enzyme provides specific environments found inside the cells. They have pocket like structure called as active sites ( is only small portion of total protein and contains substrate binding site and catalytic site. Catalytic site contains various amino acid residue which reacts with substrate and form a complex structure). Molecule is bounded by the active sites and acted upon by the enzyme is called as substrate. Most of the study done regarding the ES complex since this complex decides overall rate of reactions means formation of products. Actually an enzyme decreases the free energy between the ground state and the transition state. This difference of energy is called as activation energy. Higher activation energy reflects the slower reaction. The rate of reaction can be increases by increasing
the temperature. Catalyst or enzymes decreases this requirement by lowering down the activation energy. Enzymes never disturb the equilibrium. See graph in the figure. Transition state. ES Free energy G Enzyme lowers the activation energy. P Reaction coordinate R= reactant P = product G = H T S this is the thermodynamical term used to explain the stability of reactions. A negative free energy stabilizes the system and makes the reaction possible. H denotes the enthalpy and S denotes the entropy. In unstabilized or uncatalysed system S is positive and formation of transition complex renders them lowering of entropy leading to make overall free energy negative. A more ordered system also has lower entropy for example free amino acid has more entropy than the amino acid linked with the peptide bond. How enzymes speed up the reaction without taking part? In cells and organisms most reactions are catalyzed by enzymes, which are regenerated during the course of a reaction. These biological catalysts are physiologically important because they speed up the rates of reactions many folds (up to 10 6 to 10 15 times than normal one) that would otherwise be too slow to support life. Enzymes increase reaction rates--- sometimes by as much as one millionfold, but more typically by about one thousand fold. Catalysts speed up the forward and reverse reactions proportionately so that, although the magnitude of the rate constants of the forward and reverse reactions is are increased, the ratio of the rate constants remains the same in the presence or absence of enzyme. Since the equilibrium constant is equal to a ratio of rate constants, it is apparent that enzymes and other catalysts have no effect on the equilibrium constant of the reactions they catalyze. Enzymes increase reaction rates by decreasing the amount of energy required to form a complex of reactants that is competent to produce reaction products. This complex is known as the activated state or transition state complex for the reaction. Enzymes and other catalysts accelerate reactions by lowering the energy of the transition state. The free energy required to form an activated complex is much lower in the catalyzed reaction. The amount of energy required to achieve the transition state is lowered; consequently, at any instant a greater proportion of the molecules in the population can achieve the transition state. The result is that the reaction rate is increased. Unit of activity Micromole substrate consumed or product formed per minute is referred as IU international unit. SI unit is katal that is defined as Micromole substrate consumed or product formed per second.
Chapter 3 Enzyme Specificity Although enzymes are highly specific for the kind of reaction they catalyze, the same is not always true of substrates they attack. For example, while succinic dehydrogenase (SDH) always catalyzes an oxidation-reduction reaction and its substrate is invariably succinic acid, alcohol dehydrogenase (ADH) always catalyzes oxidation-reduction reactions but attacks a number of different alcohols, ranging from methanol to butanol. Generally, enzymes having broad substrate specificity are most active against one particular substrate. In the case of ADH, ethanol is the preferred substrate. Enzymes also are generally specific for a particular steric configuration (optical isomer) of a substrate. Enzymes that attack D sugars will not attack the corresponding L isomer. Enzymes that act on L amino acids will not employ the corresponding D optical isomer as a substrate. The enzymes known as racemases provide a striking exception to these generalities; in fact, the role of racemases is to convert D isomers to L isomers and vice versa. Thus racemases attack both D and L forms of their substrate. As enzymes have a more or less broad range of substrate specificity, it follows that a given substrate may be acted on by a number of different enzymes, each of which uses the same substrate(s) and produces the same product(s). The individual members of a set of enzymes sharing such characteristics are known as isozymes. These are the products of genes that vary only slightly; often, various isozymes of a group are expressed in different tissues of the body. The best studied set of isozymes is the lactate dehydrogenase (LDH) system. LDH is a tetrameric enzyme composed of all possible arrangements of two different protein subunits; the subunits are known as H (for heart) and M (for skeletal muscle). These subunits combine in various combinations leading to 5 distinct isozymes. The all H isozymes is characteristic of that from heart tissue, and the all M isozymes is typically found in skeletal muscle and liver. These isozymes all catalyze the same chemical reaction, but they exhibit differing degrees of efficiency. The detection of specific LDH isozymes in the blood is highly diagnostic of tissue damage such as occurs during cardiac infarct. Thus enzyme may be group specific if they act over on several closely related substrate for example alcohol dehydrogenase on which several alcohol are substrate, or act over by specific substrate like glucokinase that transfer phosphate from ATP to glucose. They exhibit absolute specificity. They may be of product specific or substrate specific or stereospecific act on specific stereo molecules like alanine recemase acts on only Lalanine to convert them into Dalanine.
Specificity is actually decided by amino acid residue present in the active site of enzyme that is capable of binding to the substrate molecule. The amino acids that are not involved in binding to the substrate do not have any contribution towards the specificity. Therefore both the subunit of enzyme like binding and catalytic site is the active center of the enzymes. They are only small part of the total enzyme and they must be accessible to the substrate. For example chymotrypisn are inactive because their zymogen form dont have accessible catalytic center. Therefore further cleavage occurs to make them active exposing the catalytic center. For proper catalytic activity their side chains must be of suitable size, shape, and character and must not block the substrate from binding. For example of several seine proteases like chymostrypsinogen, trypsin and elastases having almost similar structure but differ in specificity due to variations in the side chain amino acids. In Elastases substrate are blocked from binding due to presence of two bulky side chain valine and threonine while chymostrypsin and trypsin have no bulky group but instead contains glycine amino acid that facilitates the substrate to approach easily. Replacement of glycine by alanine make the enzyme more active. Active site most often contains both polar as well as non-polar amino acid thus facilitates the interaction with both hydrophilic and hydrophobic amino acid. This is the reason why sometime in presence of non-polar solvent like DMSO dimethylsulphoxide some enzyme are more reactive. Other examples include chymotrypsin, alcohol dehydrogenase, etc., ie. a variety of enzymes from different classes have been shown to function in nonaqueous media. Specificity May Be Very Strict Or Relatively Broad Broad specificity is shown by many degradative enzymes e.g. alkaline phosphatase, which removes phosphates from a wide range of molecules, or carboxypeptidase which snips the C-terminal amino-acid off many polypeptide chains. Intermediate specificity is most common e.g. alcohol (ethanol) dehydrogenase of E. coli will act on C2, C3, and C4 alcohols; pyruvate dehydrogenase will also use the C4 homolog of pyruvate etc.Some enzymes are absolutely specific. Aspartase catalyses the interconversion of aspartate and fumarate: L-Aspartate Fumarate + NH3 Fig . showing asymmetric binding site in the enzymes. The model of enzyme structure was proposed by fisher as three point one catalytic site and others are binding site. Sometime enzymes have both catalytic as well as binding site as same site. Such enzymes follows michaelis behaviour while others having complex and separate site behave in more complex way as allosteric site. It will only use aspartate (not similar amino acids such as glutamate) and only the L-isomer. Nor will it add NH3 to maleate, the cis isomer of fumarate.
Three theory were proposed to explain the specificity of enzyme but all of them are correct and none of them are alone operative in the enzyme specificity. 1Lock and key hypothesis 2Induced fit hypothesis. 3Transition state stabilization ENZYME MECHANISM MODELS There are two major considerations - substrate specificity and catalytic power. Enzymes must bind the correct substrate and position it correctly relative to catalytically active groups in the active site. Most enzymes are very large relative to their substrates. The actual reaction occurs in the active site, a small region of the enzyme where the substrate is bound. A. Lock and Key Model for Enzyme Activity The high specificity and efficiency of enzymes can be explained by the manner that
they associate with the reactant molecules called the substrate. One of the first theories or models that help to explain this phenomenally efficient catalytic efficiency of enzymes is called the "lock and key" model. According to this model, each enzyme molecule may have as few as one active site on the surface of the enzyme molecule itself. An active site is an indentation or cavity whereby a reactant molecule (substrate) is attached to. This is called the enzyme-substrate complex. The polar and non-polar groups of the active site attract compatible groups on the substrate molecule so that the substrate molecule can effectively lock into the cavity and position itself for the necessary collisions and bond breaks and formations that must take place for successful conversion to a product molecule. Once the product molecule has been formed the electrical attractions that made the substrate molecule adhere to the active site no longer are present, and the product molecule can disengage itself from the active site thus freeing the site for another incoming substrate molecule. This process occurs in a highly efficient manner hundreds or even thousands of times in a short time span. This model assumes that molecules that lock into the active site must form a perfect fit. Also the assumption is that the active site conformation is ridged. Evidence does not support these assumptions. For example, certain molecules can lock into the active site even though the bogus molecules have a different shape compared to the true substrate molecule. This has the effect of inhibiting enzyme activity. This does not seem to support the ridged active site assumption in the lock and key model. Furthermore, small temperature changes and small changes in pH will not result in the enzyme being inhibited from catalyzing its intended reaction. The occurrence of pH and temperature ranges of optimum enzyme activity does not support the assumptions made by the lock and key model of ridged active site cavities. Modification of the lock and key model is necessary to account for the occurrence of pH and temperature ranges of optimum enzyme activity and to explain why other molecules can effectively block the active site. B. Induced Fit (Hand and Glove) Model of Enzyme Activity Modification of the lock and key model assumes that the active site has a certain amount of elasticity whereby the active site can expand or contract in a limited way in order to accommodate the substrate molecule. The analogy is like a hand fitting into a glove. The glove adjusts in shape and size to fit various sized hands within a certain range. This tolerance would explain why bogus molecules of slightly different size compared to the true substrate molecule can still be accommodated by the elastic active site. Small changes in temperature would distort the active site conformation but not so much that the active site could not still accommodate the substrate molecular size. PH changes which would also change the active site conformation but not so much that the active site could not flexibly accommodate the substrate molecule. The Induced Fit Model seems to explain why there is some flexibility in the abilility of the active site to accommodate other molecules and at limited temperature and pH ranges. Induced fit assumes that the active site of an enzyme is not complementary to that of the transition state in the absence of the substrate. Such enzymes will have a lower value of kcat/KM (a numerical value to compare the catalytic efficiency of enzyme), because some of the binding energy must be used to support the conformational change in the enzyme. Induced fit increases KM without increasing kcat. Induced fit example Hexokinase catalyzes the ATP-dependent phosphorylation of glucose. The binding of glucose to the enzyme induces a major conformational change - the induced fit. Phosphoglycerate kinase catalyzes the synthesis of ATP. The two substrates are brought together in the active site by a conformational change. An induced fit at an interface. Lipases are enzymes that hydrolyze esters of long chain fatty acids. These enzymes show virtually no activity against water-soluble substrates and only hydrolyze substrates present in the interface between water and lipid -
interfacial activation. The activation of the lipases at the interface is a result of a conformational change, induced fit, caused by the enzyme binding to the interface. This conformational change exposes the active site of the enzyme. Mechanisms of Enzyme Catalysis Enzymes are proteins. Every protein is determined by its amino acid sequence (its primary structure) and its tertiary structure (the three-dimensional folding of the polypeptide chain). Its uniqueness is caused by the sequence and nature of its amino acid side chains. They form a number of weak interactions, which again are the basis for the spatial arrangement (conformation) of the molecule and which help to maintain this structure by stabilizing it. In that way, the onedimensional information that was stored in the genome as a DNA sequence is first transcribed into mRNA and after translation into an amino acid sequence finally transformed into a three-dimensional structure. It is this structure that allows an enzyme to perform its catalytic activity. That explains the specificity of catalysis and the selectivity for a certain substrate (and also that for additional regulatory factors). Enzyme molecules are, compared to most of their substrate molecules, rather large. Their surface is not evenly structured but displays dent-ins, grooves, pockets, hollows etc. The part that binds to a substrate molecule is termed the active centre or substrate-binding site and is characterized by a shape complementary to that of the substrate molecule. Furthermore, certain amino acid side chains are exposed at the active centre. They are engaged in the catalytic turn-over of the substrate. Numerous enzymes depend on additional factors that are necessary to perform the catalytic reaction. In other words: the enzyme binds first to a coenzyme like NAD or FAD. Often, the enzyme's surface is structured in a way that the coenzyme is bound to a specifically shaped pocket. Depending on the type of molecule, the binding is either reversible (achieved through weak interactions) or irreversible (covalent bonds). The shape of the holoenzyme (= apoenzyme [protein] + coenzyme) causes the substrate selectivity. Atoms or ionized groups of the coenzyme take part in the catalytic reaction. These properties explain, why reactions take place at an enzyme's surface that occur in solutions only after a considerable amount of activation energy has been supplied. The binding sets reactants into a state of enhanced reactivity recognizable by the close vicinity and the right orientation towards each other. The collision theory states that the event of two molecules meeting in solution in a way that a bond can form between them is rather improbable, though the probability can be considerably increased by the supply of energy (pressure, temperature).
Chapter 4 Enzyme-Substrate Interactions The favored model of enzyme substrate interaction is known as the induced fit model. This model proposes that the initial interaction between enzyme and substrate is relatively weak, but that these weak interactions rapidly induce conformational changes in the enzyme that strengthen binding and bring catalytic sites close to substrate bonds to be altered. After binding takes place, one or
more mechanisms of catalysis generate transition- state complexes and reaction products. The possible mechanisms of catalysis are five in number: 1. 2. 3. 4. 5. Catalysis by approximation General acid, general base catalysis Catalysis by electrostatic effects Covalent catalyis (nucleophilic or electrophilic) Catalysis by strain or distortion
a. Catalysis by Bond Strain In this form of catalysis, the induced structural rearrangements that take place with the binding of substrate and enzyme ultimately produce strained substrate bonds, which more easily attain the transition state. The new conformation often forces substrate atoms and bulky catalytic groups, such as aspartate and glutamate, into conformations that strain, existing substrate bonds. b. Catalysis by Proximity and Orientation Enzyme-substrate interactions orient reactive groups and bring them into proximity with one another. In addition to inducing strain, groups such as aspartate are frequently chemically reactive as well, and their proximity and orientation toward the substrate thus favors their participation in catalysis. The classic way that an enzyme increases the rate of a bimolecular reaction is to use binding energy to simply bring the two reactants in close proximity. If DG is the change in free energy between the ground state and the transition state, then DG=DHtDS. In solution, the transition state would be significantly more ordered than the ground state, and DS would therefore be negative. The formation of a transition state is accompanied by losses in translational entropy as well as rotational entropy. Enzymatic reactions take place within the confines of the enzyme active-site wherein the substrate and catalytic groups on the enzyme act as one molecule. Therefore, there is no loss in translational or rotational energy in going to the transition state. This is paid for by binding energy. c. Catalysis Involving Proton Donors (Acids) and Acceptors (Bases) Other mechanisms also contribute significantly to the completion of catalytic events initiated by a strain mechanism, for example, the use of glutamate as a general acid catalyst (proton donor). General acid-base catalysis is involved in a majority of enzymatic reactions. General acidbase catalysis needs to be distinguished from specific acidbase catalysis. Specific acidbase catalysis means specifically, OH or H+ accelerates the reaction. The reaction rate is dependent on pH only, and not on buffer concentration. In General acidbase catalysis, the buffer aids in stabilizing the transition state via donation or removal of a proton. Therefore, the rate of the reaction is dependent on the buffer concentration, as well as the appropriate protonation state. In the second step (collapse of the tetrahedral intermediate), the leaving group must be protonated. The general acidbase is best when its pKa is near that of the pH of the solution, in order to have appropriate concentrations of each buffer species. Specific acid-base catalysis is due to H+ or OH- ions which temporarly donates proton or accept proton. General acid-base catalysis is due to proton donors or proton acceptors, which donate or remove protons to (or from) the transition state intermediate. Most hydrolytic enzymes use acid/base catalysis. General acid-base catalysis is a commonly employed mechanism in enzyme reactions, e.g., hydrolysis of ester/ peptide bonds, phosphate group reactions,
addition to carbonyl groups, etc. General acid catalysis involves donation of a proton by the catalyst. General base catalysis involves abstraction of a proton by the catalyst. The side chains of Asp, Glu, His, Cys, Tyr and Lys can be involved in general acid-base catalysis. Concerted acid-base catalysis Providing both acid and base simultaneously is impossible in free solution, yet enzymes can do this. An acidic group in one part of the active site donates a proton and a basic group in another part of the active site removes a second proton from the reaction intermediate. Amino acids with extra carboxyl or amino groups can obviously act as acids or bases. In addition, histidine (pK around 7.0) can act as either an acidic or basic catalyst depending on whether or not it is protonated. Also, histidine reacts very fast (protonation/deprotonation of imidiazole ring has a reaction half-time of less than 10-10 seconds at neutral pH). Histidine is rarely found in proteins except at catalytic sites. d. Covalent Catalysis: In catalysis that takes place by covalent mechanisms, the substrate is oriented to active sites on the enzymes in such a way that a covalent intermediate forms between the enzyme or coenzyme and the substrate. One of the best-known examples of this mechanism is that involving proteolysis by serine proteases, which include both digestive enzymes (trypsin, chymotrypsin, and elastase) and several enzymes of the blood clotting cascade. These proteases contain an active site serine whose R group hydroxyl forms a covalent bond with a carbonyl carbon of a peptide bond, thereby causing hydrolysis of the peptide bond. Biologically important nucleophiles are negatively charged or contain unshared electrons Biologically important electrophiles generally are either positively charged, or contain unfilled valence electron shells, or contain an electronegative atom Covalent Intermediates Here the strategy is to lower the transition state energy "hump" by taking an alternative reaction pathway therefore sometime it is also called as alternative pathway catalysis. Sometime nucleophilic catalyst form intermediate more rapidly than normal catalysis. In covalent catalysis, a covalent bond is transiently formed between the substrate and the enzyme (or coenzyme). This reaction usually involves a nucleophilic group on the enzyme and an electrophilic group on the substrate Uncatalysed: BX + Y BY + X Catalysed: BX + Enz Enz-B + X Enz-B + Y Enz + BY Where, B is usually some chemical group such as a phosphate group, acyl group or glycosyl group. Phosphoenzymes usually attach the phosphate to serine or histidine. Serine type - phosphoglucomutase, alkaline phosphatase. Histidine type - glucose 6 phosphatase, phosphotransferase system. Acyl enzymes usually attach the acyl group at an active serine or cysteine residue. Most proteases (eg trypsin, elastase, subtilisin are serine enzymes. Cysteine enzymes include papain (a protease), glyceraldehyde phosphate dehydrogenase and most enzymes using acyl CoA derivatives. The proteases are globular, water soluble proteins that function as enzymes. They catalyze the hydrolyses of peptide bonds in proteins. Being enzymes, proteases can be characterized by their substrate affinity and the catalytic rate of the
reaction. Using proteases to study the effects of single amino acid substitutions (mutations) on catalytic rate and substrate affinity demonstrated that these two properties are linked and that this linkage can be explained by analyzing the conformation of the catalytic or active site of the enzyme. This analysis showed that four major functional groups are found in the catalytic site of proteases and based on this functional groups, 4 families of proteases have been defined: a. Serine proteases b. Cystein proteases c. Aspartate proteases d. Metallo proteases This classification uses the functional group within the enzyme, and does not relate to the substrate specificity of the proteases themselves. The name of the protease family refers to an amino acid (e.g. aspartate) or a metal as co-enzyme at the active site of the enzyme. Structural element of active site Function The main chain substrate binding unspecific binding of polypeptide segment Specificity pocket semi-specific binding of side chains, sequence specificity Oxyanion whole stabilizes S* over S in enzyme The catalytic triad forms tetrahedral intermediate (transition state; stabilizes S* over S); hydrolyzes peptide bond The serine proteases are a class of enzymes that degrade proteins in which a serine in the active site plays an important role in catalysis. The family includes among many others, Chymotrypsin and trypsin, which weve talked about, and Elastase. All three enzymes are similar in structure, and they all have three important conserved residuesa histidine, an aspartate, and a serine. Thrombin is crucial in the blood-clotting cascade Subtilisin is bacterial protease Plasmin breaks down the fibrin polymers of blood clots Tissue plasminogen activator (or TPA), cleaves the proenzyme plasminogen, yielding plasmin The six step reaction mechanism of chymotrypsin: 1. Substrate attaches to enzyme at the main chain binding site and specificity pocket with the scissile (peptide) bond exposed to the triad (ES formation) 2. covalent bond formation between -C=O and S195 hydroxyl (nucleophilic attack), tetrahedral intermediate as transition state, oxyanion hole stabilization, H57 binds the -released H+ from S195 (ES*) 3. acyl-enzyme intermediate and peptide bond hydrolysis, peptide with new N-term released from enzyme taking -H from H57 (originally S195; see step 2) (EP*) 4. A water molecule placed next to H57 and acyl-enzyme intermediate at S195 (EP*) 5. the water molecule initiates a nucleophilic attack and undergoes a reaction with one -H attaching to H57 and -OH to acyl-enzyme intermediate forming again a tetrahedral transition state of the new C-terminal peptide fragment (EP*) 6. Peptide released while H57 donates -H (from water molecule) to S195 (E+P) Chymotrypsin cleaves after mainly aromatic amino acids, while trypsin cleaves after basic amino acids. Elastase is fairly nonspecific, and cleaves after small neutral amino acids. Notice how their active sites are suited for these tasks. Chymotrypsin is a protease that cleaves peptide bonds on the carboxyl side of aromatic or large hydrophobic amino acids the mechanism of chymotrypsins cleavage reaction includes covalent, general acid-base, electrostatic, and proximity/orientation catalysis The tetrahedral intermediate in chymotrypsin, which consists of the Ser195 adduct before departure of the leaving group, is considered to be the transition
state intermediate in the chymotrypsin reaction. It is high energy because there is a carbon surrounded by 3 electronegative atoms, one of which bears a negative charge. How is it that the enzyme stabilizes this transition state intermediate? The backbone amides of gly193 and ser195 form an oxyanion hole. They loosely hydrogen bond to the carbonyl oxygen under attack. Upon formation of the tetrahedral intermediate, the resulting carbon-oxygen single bond is longer, and the negatively charged oxygen is better accommodated in the oxyanion hole. Diisopropylflurophosphate is an inhibitor of chymotrypsin. It diffuses into the active, wherein a nucleophilic amino acid attacks the phosphate, releasing fluoride anion. This results in a covalent bond between the nucleophile and the inhibitor. It inhibits the reaction because it blocks entry of normal substrates. The enzyme-inhibitor adduct is very stable. Upon hydrolysis of the protein (6 N HCl, 110C) and amino acid analysis on the hydrolysate, a novel amino acid was isolated. It was the diisopropylphosphoryl derivative of serine. Crystal structures of chymotrypsin reveal the reason for the high reactivity of Ser 195 Ser 195 is in a H-bonding interaction with His 57, which is H-bonding with Asp 102 (catalytic triad) the catalytic triad serves to activate Ser 195 through general acid-base catalysis catalytic triads are common in serine proteases Conformation changes from trigonal to tetrahedral in the peptide upon nucleophilic addition allow the oxyanion formed to move into a hole with two backbone amides inside the oxyanion hole the transition state anion can form two hydrogen bonds with the amide hydrogens as donors the enzyme cannot form H-bonds with the amide hydrogens in the oxyanion hole when the peptide is in trigonal conformation The mechanism of chymotrypsin utilizes stabilization of the transition state, covalent catalysis, acid-base catalysis, electrostatic, proximity and orientation effects Substrate specificities in proteases are due to active site binding pockets. Chymotrypsin substrate specificity is due almost entirely to one deep, hydrophobic pocket that binds the sidechain of the amino acid N-terminal to the site of bond cleavage (S1) binding of appropriate sidechains to this pocket positions the adjacent peptide bond into the active site for cleavage Many serine proteases have high similarity to chymotrypsin trypsin and elastase are approximately 40% identical in primary sequence to chymotrypsin and their overall structures are very close Substrate specificity of these other proteases compared to chymotrypsin can be attributed to their S1 binding pockets Trypsin: cleaves after positively charged residues, S1 has Asp Elastase: cleaves after small side chains (A, S), S1 has bulky Val residues
Histidine, serine and cysteine operate by nucleophilic catalysis. Their nucleophilic groups (imidazole ring, OH and SH respectively) are good electron donors the intermediates they form are unstable and react easily with the final acceptors. Since the whole point is that covalent intermediates should rapidly break down to release the reaction product, enzyme covalent intermediates are very difficult to isolate.
cystein prtoease Renin - An Aspartyl Protease A Metalloprotease with Substrate Carbonic Anhydrase Mechanism His64 assists in H+ removal HCO3- released. H2O enters HO- attacks CO2
Methylation prevents H-bonding with DNA substrate Lysozyme is a small globular protein composed of 129 amino acids. It is also an enzyme which hydrolyzes polysaccharide chains, particularly those found in the peptidoglycan cell wall of bacteria. In particular, it hydrolyzes the glycosidic bond between C-1 of N-acetyl muramic acid and C-4 of Nacetyl glucosamine. It is found in many body fluids, such as tears, and is one of the bodys defenses against bacteria. The best studied lysozymes are from hen egg whites and bacteriophage T4. Although crystal structures of other proteins had been determined previously, lysozyme was the first enzyme to have its structure determined. The X-ray crystal structure of lysozyme has been determined in the presence of a nonhydrolyzable substrate analog. This analog binds tightly in the enzyme active site to form the ES complex, but ES cannot be efficiently converted to EP. It would not be possible to determine the X-ray structure in the presence of the true substrate, because it would be cleaved during crystal growth and structure determination. The active site consists of a crevice or depression that runs across the surface of the enzyme. Look at the many hydrogen bonding contacts between the substrate and enzyme active site that enables the ES complex to form. There are 6 subsites within the crevice, each of which is where hydrogen bonding contacts with the sugars are made. In site D, the conformation of the sugar is distorted in order to make the necessary hydrogen bonding contacts. This distortion raises the energy of the ground state, bringing the substrate closer to the transition state for hydrolysis. At what position does water attack the sugar? When the lysozyme reaction is run in the presence of H218O, 18O ends up at the C-1 hydroxyl group at site D. This suggests that water adds at that carbon in the mechanism. From the X-ray structure, it is known that the C-1 carbon is located between two carboxylate residues of the protein (Glu-35 and Asp-52). Asp-52 exists in its ionized form, while Glu-35 is protonated. Glu can act as a general acid to protonate the leaving group in the transition state. Asp can function to stabilize the positively charged intermediate. Glu then acts as a general base to deprotonate water in the transition state. Within the class of hydrolases, Lysozyme belongs to the Glycosylases family (EC 3.2.-.-). Lysozyme reaction is the hydrolysis of the beta (1-4) glycosidic bond between N-acetylglucosamine sugar (NAG) and N-acetylmuramic acid sugar (NAM) and therefore it is possible classify it as Glycosidases, i.e. enzymes hydrolyzing O- and S-glycosyl (EC 3.2.1.-) with number 17 (EC 3.2.1.17) in this group
Lysozyme Lysozyme catalyzes the hydrolysis of the (1-->4) linkage between N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) in bacterial cell wall polysaccharides and (1 4)-linked poly NAG (chitin). The lysozyme mechanism illustrates: Transition state stabilization Acid-base catalysis Several residues in the protein participate in substrate binding. The binding of NAM4 in the chair conformation is unfavorable. But the binding of residue 4 in the half-chair conformation is favorable (preferential transition state binding). Hydrolysis involves acid-base catalysis (Glu35 serves as a proton donor to the oxygen of the leaving alcohol. The resulting carbonium ion (+) is stabilized by the ionized side chain of Asp52 until it can react with water) Many glycosidases utilize a covalent intermediate in their mechanism. Here is a good example of transition state stabilization using a bacterial glycosidase. Another excellent example that clearly shows how transition state stabilization works is found in the structure of this transferase. Chloroketones are inhibitors of serine proteases, which bind in the active site and react with His 57. Pancreas contains a small protein (6 kDa) that is a potent inhibitor of trypsin. The inhibitor is a naturally occurring transition state analog of a protein substrate. e) Metal Ion Catalysis Nearly 1/3 of all known enzymes require the presence of metals for catalytic activity Metalloenzymes - contain tightly bound metal cofactors such as Fe2+, Fe3+, Cu2+, Zn2+, Mn2+, Co2+ Metal Activated enzymes only loosely bind the metal ions. The ions are usually Na+, K+, Mg2+, or Ca2+ Many enzymes have metal ions at the active site. Sometimes these are used as redox centers, as in cytochromes, nitrate reductase etc. However, often the metal ion does not get reduced or oxidized, but acts to stabilize negative charges on the reaction intermediate. In carboxypeptidase, Zn2+ polarizes the C=O of the peptide bond which is about to be broken. In alcohol dehydrogenase Zn2+ polarizes the C=O
group of acetaldehyde, so allowing the hydride ion (H) to be added to the carbon atom. Electrostatic interactions are much stronger in organic solvents than in water due to the dielectric constant of the medium. The interior of enzymes have dielectric constants that are similar to hexane or chloroform. Metal ions that are bound to the protein (prosthetic groups or cofactors) can also aid in catalysis. In this case, Zinc is acting as a Lewis acid. It coordinates to the non-bonding electrons of the carbonyl, inducing charge separation, and making the carbon more electrophilic, or more susceptible to nucleophilic attack. Metal ions can also function to make potential nucleophiles (such as water) more nucleophilic. For example, the pka of water drops from 15.7 to 6-7 when it is coordinated to Zinc or Cobalt. The hydroxide ion is 4 orders of magnitude more nucleophilic than is water. SUMMARY Metal Mn+2. Ca+2. ion catalysis is widely used. Binding substrates in the proper orientation. Mediating oxidation-reduction reactions. Electrostatically stabilizing or shielding negative charges. Metalloenzymes contain tightly bound metal ions: Fe+2, Fe+3, Cu+2, Zn+2, Metal-activated enzymes contain loosely bound metal ions: Na+, K+, Mg+2,
Electrostatic catalysis refers to the fact that when a substrate binds to an enzyme, water isusually excluded from the active site. This causes the local dielectric constant to be lower, Which enhances charge-charge interactions in the active site. Proximity and orientation effects are important in enzymatic reactions. The three dimensional structure of the enzyme can bring several reactive side chains into close proximity in the active site. Binding of the substrate in the active site can orient the substrate for most efficient interaction with these side chains. Preferential Transition State Binding is probably the most important rate enhancing mechanism available to enzymes. This means that the enzyme binds the transition state of the reaction more tightly than either the substrate or product, therefore Go is lowered. Weak interactions between the enzyme and substrate are optimized in the transition state. Utilization of enzyme-substrate binding energy in catalysis The maximum binding energy between an enzyme and a substrate will occur when there is maximal complementarity between the structure of the binding site and the structure of the substrate. The structure of the substrate changes during the reaction, becoming first the transition state and then products. The structure of the enzyme can only be complementary to one form of the substrate. If the structure of the active site is complementary to the transition state, then binding increases as the reaction proceeds, which lowers the activation energy. Having the active site bind the transition most strongly also means that
products are bound weakly, which favors dissociation of the products from the enzyme once the reaction is complete. When the active site is complementary to the transition state the full substrate binding energy is only realized as the reaction reaches the transition state, which lowers the activation energy of the reaction by the magnitude of the binding energy. Molecular Mechanisms for the Utilization of Binding Energy Strain is a classic concept in which it was supposed that binding of the substrate to the enzyme somehow caused the substrate to become distorted toward the transition state. Transition state stabilization is a more modern concept, which states that it is not the substrate that is distorted but rather that the transition state makes better contacts with the enzyme than the substrate does, so the full binding energy is not achieved until the transition state is reached. Strain or stress? It is unlikely that there is enough energy available in substrate binding to actually distort the substrate toward the transition state. It is possible that the substrate and enzyme interact unfavorably and this unfavorable interaction is relived in the transition state. We might think that the substrate is under stress meaning that it is subjected to forces but not distorted by them. It is more likely that the enzyme is strained, as for example in induced fit.
Catalytic Power Enzymes lower the energy of the transition state by stabilizing the original reaction intermediate or by providing an alternative reaction pathway. Rate increases by enzymes range from 108 to 1020 relative to the uncatalysed, spontaneous reaction (e.g. 109 for alcohol dehydrogenase; 1016 for alkaline phosphatase). Factors involved in enzyme rate increases: a) Proximity - up to 106 fold; b)Orientation - up to 100 fold c) Covalent enzyme-substrate intermediates around 1010 fold d) General acid-base catalysis - around 1010 fold ;e) Metal ion catalysis - around 1010 fold; f) Distortion of the substrate - up to 108 fold (but largely hypothetical) a) Proximity The enzyme binds the substrate so that the susceptible bond is very close to the catalytic group in the active site or in close proximity in the active site. It increases their concentration and and reduces entropy loss for subsequent formation of a transition state. This has been called as proximation effect or approximation effect. Therefore it appears that enzyme bound transition state is the single most in determining whether the reaction should proceed. Calculations indicate that the local concentration of substrate in the active site may be as much as 50M whereas its concentration in the cytoplasm may be less than 1mM. Since chemical reaction rates are proportional to the concentrations of the reactants a rate enhancement of up to 106 may be generated by local concentration effects. This effect is counteracted to some extent by the fact that the concentration of enzyme active sites is low (the active site is a small portion of a very large molecule; the total protein concentration in cytoplasm is 1 to 10mM at most). b) Orientation Proximity alone is insufficient. The reacting groups must be properly oriented. Orbital steering hypothesis - binding of the substrate(s) to the enzyme aligns the reactive groups so that the relevant molecular orbitals overlap. This increases
the probability of forming the transition state. For a typical bimolecular reaction in free solution about 1/100 collisions between molecules of sufficient energy actually leads to a reaction. Thus the maximum effect of orientation would be 100-fold. Consider the attack on an aryl ester by a carboxylic acid. Now let's put both reacting groups on the same molecule - i.e. we will link R1 and R2 together. As the five examples show, the relative rate gets greater as the reacting groups are brought closer together. In addition, holding them in the correct orientation also helps. (Ar = methoxyphenyl group.)
f. Catalysis in organic solvents Another approach that has generated some interest in enzyme technology is catalysis in non-aqueous or nearly anhydrous media (Bell et al. 1995; Carrea et al. 1995; Gupta, 1992). Alexander Klibanov & his colleagues at MIT initiated much of the work in this area. Traditionally, enzyme reactions are carried out in aqueous media. The question arises as to how much water is really needed by the enzyme to function. This led them to test how enzymes function in various amount of organic solvents. They found startling changes to enzyme properties as the water content of the reaction mixture decreases. Some of these are illustrated in overheads for porcine pancreatic lipase (Zaks & Klibanov 1984; 1985). They concluded that it is the water bound to the enzyme that is critical for activity, rather than the total water content of the system. Research by Klibanov, his colleagues and other researchers have opened up novel opportunities in enzyme technology and this field has changed from a mere curiosity to a serious alternative to chemical reactions. The functions of a number of enzymes under non-aqueous conditions have been studied. Such systems are applicable to enzymes of varying reaction types. Some examples include: 1. Lipases, which show increased thermostability, altered substrate specificity and a propensity towards synthetic reactions in non-aqueous media. 2. Horse radish peroxidase (HRP) which is able to depolymerize lignin in 95% dioxane (Dordick et al., 1986). 3. Glucose isomerase for conversion of glucose to fructose to produce high fructose corn syrup (HFCS). 4. Other examples include chymotrypsin, alcohol dehydrogenase, etc., ie. a variety of enzymes from different classes have been shown to function in nonaqueous media. Some advantages to using enzymes in non-aqueous solvents include (Dordick, 1989): 1. Increased solubility of nonpolar substrates. 2. Shifting thermodynamic equilibria to favor synthesis over hydrolysis. 3. Reduction in water-dependent side reactions. 4. Immobilization is unnecessary, because enzymes are insoluble in organic solvents, and can be recovered by simple filtration. 5. Ease of product recovery from low boiling organic solvents. 6. Enhanced thermo stability of enzymes; water is required to inactivate enzymes at high temperature. Therefore, in the absence of water, enzymes are more stable. 7. Altered activity and affinity (Km) of the enzyme. 8. Novel enzyme reactions possible (eg. new catalytic activity seen with lipases or horse radish peroxidase). 9. Elimination of microbial contamination. 10. Can use enzymes directly within a chemical process. Importance and relevance of whole cell enzymes in biotechnology Enzymes are biological catalysts. With the exception of ribozymes, enzymes are
mainly proteins in nature, and may also possess lipid or sugar moieties. Their main task is to decrease the activation energy (Eact) of a chemical reaction. The whole biotechnology field depends on the activities of enzymes, either acting alone or in concert. Exploitation of enzymes is not a recent development. They have been used throughout the ages in leather tanning, in cheese-making, in the preparation of malted barley for beer brewing & the leavening of bread. These processes use enzymes in the form of whole cells. Conceptually, it is easy to decide whether one should use whole cells or isolated enzymes in a biotechnological process to produce or transform compounds. Some processes require multi-step transformations, eg. the synthesis of antibiotics, interferons, monoclonal antibodies, or ethanol production from glucose. These transformations involve a number of enzymes acting sequentially, along with the requirement for co-factor regeneration in several steps. They are clearly candidates for using whole cells. There may be some drawbacks to using whole cells in catalysis. These include 1. Competing side reactions from other enzymes. 2. Sterility problems often arise when one deals with whole cells. 3. Some conditions may lead to cell lysis, and this can result in loss of biocatalysts or severely decrease reaction rate depending on the nature and type of enzyme reaction. 4. Formation of by-products. 5. On the other hand, with one-step or two-step stereospecific transformations, enzymes can be superior because their use will eliminate some of the potential problems with using whole cells. The advantages of using enzymes include: 6. High catalytic rate possible. 7. Functions in moderate conditions. 8. Non-polluting catalysis. 9. Less by-products formed. 10. Less problem with sterility. 11. Optimizing 1 or 2 enzyme catalyzed reactions is easier than optimizing the whole pathway in a whole cell. 12. Enzymes catalyze a wide range of reactions, some with high stereospecificity, such as oxidation, reduction, dehydrogenation, dehalogenation, hydration, dehydration, etc. Therefore, the scope of enzyme technology is very broad. 13. Despite all these striking characteristics, enzyme have not been used widely as compared to chemical catalysis. This is because the use of enzymes also suffers from some serious drawbacks: 14. Most isolated enzymes are not stable enough for use under industrial conditions. This may include poor stability with respect to temperature, pH, the presence of metal ions, etc. This is not surprising considering that most enzymes have evolved to function in a biological milieu where the conditions may differ from those used in industry. 15. In cells, all enzymes are subject to turnover, ie. old ones are replaced by new ones. This is difficult to do with isolated enzymes in vitro. 16. Most enzymes are water-soluble, and therefore, may be difficult to separate from reactants or products. 17. Many useful enzymes are intracellular, and hence are difficult to isolate. Abzymes Catalytic antibodies are also known as abzymes, are capable of catalyzing specific chemical reactions. They are able to perform this task because they have been elicited against antigen known as transition state analogues, these are molecules that closely resemble in shape and charge the reaction transition state (the highest energy spices in reaction pathway known as activated complex). Enzyme observed to bind transition complex but not the substrate, and the product. Enzyme by forming the active site similar to the transition state, enzyme stabilizes or
lowers down the energy of this species. The effect is to accelerate the reaction because the activation barrier is more easily overcome. This complementarity in shape and charge to the transition state is readily demonstrated by the effectiveness of transition state analogue is a phosphonate containing molecule that mimics the transition state of the hydrolysis of the corresponding acyl derivative. When an ester is hydrolysed the central carbonyl group changes from planer sp2 structure to tetrahedral sp3. the phosphonate containing molecule analogue is able to reproduce the shape of the transition state as well as the partially negative charged oxygen. In addition, it is chemically stable, where as the actual transition state exists only fleetingly. In this example, an enzyme that catalysed the hydrolysis of this ester would also bind tightly to phosphonate analogue. The general strategies behind the production of catalytic antibody is to (i) design and synthesize a molecule whose shape is closely resemble to that of transition state of the reaction one wishes to catalyze, (2) tether this molecule to larger molecule, (3) elicit an immune response to this conjugate, (4) screen the resultant monoclonal antibodies for catalytic activity of the type desired. Antibodies that are elicited against transition state analogue and therefore have the ability to bind them will be chemically and sterically complementary to the transition state and will therefore be potentially capable of catalyzing the reaction.
Table 7. 1 Exercises LONG TYPE 1. Describe the six classes of enzyme with examples? 2. Define Biocatalyst ? Describe how biocatalyst lower down the activation energy? 3. Define Enzyme ? Classify the Enzyme on the basis of enzyme commission? 4. Define holoenzyme ? write the role of apoenzyme, cofactor, and metalloenzymes on enzyme activity. 5. Define enzyme specific activity? Write the different method of enzyme substrate interaction in brief? Short type 1. write short notes on following a. Enzyme activity b. Enzyme specificity c. Enzyme commission. d. Holoenzyme. e. Cofactor f. Metalloenzymes. g. Coenzyme.
Module II Specificity of enzyme action, monomeric and oligomeric enzymes, Enzyme inhibition.
Chapter-5 ENZYME KINETICS & INHIBITION TERMS Enzymes are protein catalysts of biological origin that, like all catalysts, speed up the rate of a chemical reaction without being used up in the process. They achieve their effect by temporarily binding to the substrate. Competitive inhibition, when the substrate and inhibitor compete for binding to the same active site. Noncompetitive inhibition, when the inhibitor binds somewhere else on the enzyme molecule reducing its efficiency. Enzyme kinetics The study of the rate at which an enzyme works is called enzyme kinetics. Alloenzymes. If a gene exists in a heterozygous state, two different polypeptide chains will be generated. They may be separated by gel electrophoresis under favourable conditions. They may also differ in their rate of substrate turnover. The gene product a, for example, may be inactive while that of A is fully active. But all states in between are also possible. Furthermore, it was shown in numerous examples that A may be favourable under certain environmental conditions while a, is favourable under other conditions (more in the section about evolution). Polypeptides (enzymes) that are encoded by different alleles are called alloenzymes. Isoenzymes. By duplication of the genetic material a gene may be present within a haploid genome for two or more times. It is not important whether the gene loci are on one chromosome or distributed over several chromosomes. The gene products (enzymes) of the different gene loci (pseudoalleles) are called isoenzymes. Competitive inhibitors are molecules that bind to the same site as the substrate preventing the substrate from binding as they do so - but are not changed by the enzyme. Noncompetitive inhibitors are molecules that bind to some other site on the enzyme reducing its catalytic power. INTRODUCTION Kinetics is the study of the rate of change of reactants to products .Velocity refers to the change in concentration of substrate or product per unit time. Rate refers to the change in total quantity per unit time. Initial velocity is the change in reactant or product concentration during the linear phase of a reaction. Study of rate of the catalyzed reaction is called as kinetics. This is the important step in understanding the mechanism of catalysis. Therefore, it is necessary to study the factors affecting the rate of catalysis. The rate of reactions depends on the order and molceulcarity. When enzyme concentration is small and substrate concentration increases then first the reaction occurs rapidly but maximum rate is achieved only when all the space in the active site is filled
and no more substrate can be filled. This time reaction rate become slower and the product formation starts. Therefore initially the order is first and later on it becomes zero order. Therefore maximum velocity is affected by the occupying all space of the active site by the substrate. If this is occupied by the inhibitor as in competitive inhibition the v max remain same since space is same and inhibitor has similar structure as the substrate. Initially substrate consumes and product is released. This assumption is true for single substrate and single enzyme. Rate in the itegarl form is shown by the rate = dX/ dt. Chemical Reactions and Rates According to the conventions of biochemistry, the rate of a chemical reaction is described by the number of molecules of reactant(s) that are converted into product(s) in a specified time period. Reaction rate is always dependent on the concentration of the chemicals involved in the process and on rate constants that are characteristic of the reaction. For example, the reaction in which A is converted to B is written as follows: A B----------------------------1
The rate of this reaction is expressed algebraically as either a decrease in the concentration of reactant A: -[A] = k[B]-----------------------2 or an increase in the concentration of product B:
[B] = k[A]--------------------3 In the second equation (of the 3 above) the negative sign signifies a decrease in concentration of A as the reaction progresses, brackets define concentration in molarity and the k is known as a rate constant. Rate constants are simply proportionality constants that provide a quantitative connection between chemical concentrations and reaction rates. Each chemical reaction has characteristic values for its rate constants; these in turn directly relate to the equilibrium constant for that reaction. Thus, reaction can be rewritten as an equilibrium expression in order to show the relationship between reaction rates, rate constants and the equilibrium constant for this simple case. The rate constant for the forward reaction is defined as k+1 and the reverse as k-1. At equilibrium the rate (v) of the forward reaction (A B) is by definition is equal to that of the reverse or back reaction (B A), a relationship which is algebraically symbolized as: V forward = v reverse---------------4 Where, for the forward reaction: V forward = k+1[A]---------------5 and for the reverse reaction: V reverse = k-1[B]----------------6 In the above equations, k+1 and k-1 represent rate constants for the forward and reverse reactions, respectively. The negative subscript refers only to a reverse reaction, not to an actual negative value for the constant. To put the relationships of the two equations into words, we state that the rate of the forward reaction [v forward] is equal to the product of the forward rate constant k+1 and the molar concentration of A. The rate of the reverse reaction is equal to
the product of the reverse rate constant k-1 and the molar concentration of B. At equilibrium, the rate of the forward reaction is equal to the rate of the reverse reaction leading to the equilibrium constant of the reaction and is expressed by: [B]/[A] = k+1/k-1 = K eq.---------------7 This equation demonstrates that the equilibrium constant for a chemical reaction is not only equal to the equilibrium ratio of product and reactant concentrations, but is also equal to the ratio of the characteristic rate constants of the reaction. Chemical Reaction Order Order of reaction is refers to the number of molecules involved in forming a reaction complex that is competent to proceed to product(s). Empirically, order is easily determined by summing the exponents of each concentration term in the rate equation for a reaction. A reaction characterized by the conversion of one molecule of A to one molecule of B with no influence from any other reactant or solvent is a first-order reaction. The exponent on the substrate concentration in the rate equation for this type of reaction is 1. A reaction with two substrates forming two products would a second-order reaction. However, the reactants in second- and higher- order reactions need not be different chemical species. An example of a second order reaction is the formation of ATP through the condensation of ADP with orthophosphate: ADP + H2PO4 FIGURE 10. 1 FIGURE 10. 2 For this reaction the forward reaction rate would be written as: V forward = k1 [ADP][H2PO4]---------------9 L. Michaelis and M. Menten in 1913 postulated that enzyme first combine reversibly with the substrate to form an enzyme substrate complex in a relatively fast reversible step and then product is released along with free enzyme. As we know at fixed enzyme concentration, when substrate conc. Is increased then initially rate become faster and at maximum velocity where enzyme saturates the rate become steady as shown in figure. The Michaelis derived an equation to explain the fact as Initial velocity Michaelis-Menton constant In typical enzyme-catalyzed reactions, reactant and product concentrations are usually hundreds or thousands of times greater than the enzyme concentration. Consequently, each enzyme molecule catalyzes the conversion to product of many reactant molecules. In biochemical reactions, reactants are commonly known as substrates. The catalytic event that converts substrate to product involves the formation of a transition state, and it occurs most easily at a specific binding site on the enzyme. This site, called the catalytic site of the enzyme, has been evolutionarily structured to provide specific, high-affinity binding of substrate(s) and to provide an environment that favors the catalytic events. The ATP + H2O---------8
complex that forms when substrate(s) and enzyme combine is called the enzyme substrate (ES) complex. Reaction products arise when the ES complex breaks down releasing free enzyme. Between the binding of substrate to enzyme, and the reappearance of free enzyme and product, a series of complex events must take place. At a minimum S, an ES complex must be formed; this complex must pass to the transition state (ES*); and the transition state complex must advance to an enzyme product complex (EP). The latter is finally competent to dissociate into product and free enzyme. The series of events can be shown thus: E + S ES P-------------------10 and K1 k-1 The ES complex then breaks down in a slower second step to yield the free enzyme and the reaction product P: k2 ES -------- E+P ..(12) Since the second step is slower and therefore it limits the rate of overall reaction. It means that, overall rate of enzyme-catalyzed reaction must be proportional to the concentration of species that reacts in the second step, which is ES. At any time the enzyme-catalyzed reaction, the enzyme exists in the combined form E or uncombined form ES. At low [S], most of the enzyme will be in uncombined form E. here the rate will be proportional to the [s] because the equilibrium of equation will shift for the formation of more ES as S is increased. The maximum initial rate is observed when the entire enzyme E0 is present as ES complex and the concentration of E is very small. Under these state the enzyme is saturated with its substrate, so that further increase in [S] have no effect on rate. This is the condition when [S] is sufficiently high that essentially all the free enzyme is converted into the ES form. After the ES complex breaks down to yield product P the enzyme is free to catalyze another reaction. a. Pre-steady state:-The duration in which ES formation takes place is called as pre-steady state. It is too difficult to observe because of very short time. b. Steady state:-Here ES complex is constant over a period of time. Derivation: E+S k-1 k1 k2 ES E+P (13) E+S ES . 11 ES* EP E +
VO can be determined by the breakdown of ES to give product, which is determined by the [ES]: V0=k2 [ES]-----------------------------------------------------(14) TOTAL enzyme concentration can be represented by [Et]. So free enzyme can be represented by [Et][ES]. Also, because [S] is greater than Et, the amount of substrate bound by the enzyme at any given time is negligible compared to total [S]. K1 denotes the rate of formation of ES= k1( [Et][ES] [S] .. (15) k-1 denotes the rate of ES breakdown= k-1[ES] + k2 [ES]-------------------------(16)
So in the steady state, eq. 5=6 k1( [Et][ES] [S] [ES]-----------------------(17) or k1[Et][S]--- K1[ES] [S] [ES]----------------------------(18) = = k-1[ES] + k2
(k-1 + k2)
To Simplify the equation add the term k1[E] [S] to both side, we finally get K1 [Et][S] k2)[ES]-------------------------(19) Solving for the ES we get. K1[Et][S] [ES] = --------------------------------(20) or [ES] = --------------------------------(21) -----------------k1[S] + k-1+ k2 [Et][S] -----------------[S] +( k-1+ k2)/ k1 The term ( k-1+ k2)/ k1 is called as MichaelisMenten equation, the rate equation of one substrate, enzyme catalyzed reaction and is denoted by the Km. this defines the rate of initial velocity and maximum velocity. Putting value of ES in equation 21 from equation 13 gives new final equation that is Vo= V max . [S] / Km + [S]-------------------22 = (k1 [S]+k-1+
What will happen if the initial velocity is exactly one-half of maximum velocity? So if V0 = Vmax/2, then Vmax/2= V max [S]/ km+[S],-----------------23 = [S]/ km+[S] solving for km, we get Km+[S] = 2[S], or --------------------24 This represent that Km is equivalent to the substrate concentration at which Vo is one half Vmax. Note that Km has unit of molarity. This shows that the MichaelisMenten constant equals the substrate concentration at half-maximal reaction velocity. Consequently, the dimension of kM is Mol. The smaller the value of kM , the higher is the enzyme's affinity for its substrate. See figure 10.3. Km is (roughly) an inverse measure of the affinity or strength of binding between the enzyme and its substrate. The lower the Km, the greater the affinity (so the lower the concentration of substrate needed to achieve a given rate). Note :for the Michaelis-Menten reaction, k2 is the rate limiting ; thus k2<<k-1 So Km reduces to Km=k-1/k1, and it is defined as dissociation constant, Ks for the ES complex. 1. 2. Km can be very complex in the multistep enzyme catalysis. Km can change from one enzyme to other.
3. Km denotes the affinity of enzyme, but it does not tell anything about the nature of enzyme. 4. An enzyme that acts on very low concentration of substrate has very low Km. high value of Km denotes low enzyme activity. 5. Low Km enzyme can efficiently covert very low amount of substrate into the product such enzyme is said to have high affinity of enzyme while, other enzyme that requires large amount of substances for their activation and conversion into product is said to have as High Km value but low affinity. This property is efficiently utilized by the LDH enzyme that has different Km value in the heart and muscle. In muscle LDH have High Km, while in the heart it has low Km essential for the survival. On writing the Michaelis Menten constant in reciprocal form 1 / v = (kM / vmax) x 1 / [S]--------------------25 This representation is also known as the Lineweaver-Burk equation. Its advantage is in the favourable graphic depiction of the quantities. A double recessive depiction yields a straight line with a gradient of kM / vmax. This plot has advantage that it allows more accurate determination of Vmax. See figure 10.4 & 10.5 1 / kM and 1 / vmax are the points of intersection with the co-ordinates. FIGURE 10. 3 showing rate of reaction and Km at Vmax Figure 10. 4 Graphical method to find the value of Km by plotting the graph between the 1/Vo verses 1/[S] 10.4 Catalytic efficiency and Turnover number. Catalytic efficiency is denoted by the ratio of Kcat/ Km. K cat is the turn over number of enzyme . See from the table below the enzyme having very high value of Kcat have high capacity to convert substrate into the product The turnover number is a measure of catalytic activity. The kcat is a direct measure of the catalytic production of product under saturating substrate conditions.l kcat, the turnover number, is the maximum number of substrate molecules converted to product per enzyme molecule per unit of time. According to M-M model, k cat = Vmax /Et. Values of k cat range from less than 1/sec to many millions per sec. in multistep enzymatic reaction, one step may be rate limiting that rate limiting step is equivalent to Kcat. Equation 22 can be written as by placing the value of Vmax = Kcat x Et -------------------26 figure 10. 5 showing turn over number of different enzyme. Note that high the TON of an enzyme, best is the enzyme for maximum conversion of substrate into the product. FIGURE 10. 6 The constant Kcat has unit S-1, reciprocal of time. Note that two enzymes catalyzing different reaction may have the same Kcat value (turnover number) yet the rate may be different. It also denotes about the reaction between the enzyme and substrate since at low K cat, Km value will also be unsatisfactory. From equation 26, Vo depends on both Et and S, therefore K cat /Km is a second order rate constant. Therefore Kcat /Km is the best way to compare the catalytic efficiency of the enzyme. Many enzymes have K cat /Km value in the range 108 to 109 . FIGURE 10. 7
The kinetics of simple reactions like that above was first characterized by biochemists Michaelis and Menten. The concepts underlying their analysis of enzyme kinetics continue to provide the cornerstone for understanding metabolism today, and for the development and clinical use of drugs aimed at selectively altering rate constants and interfering with the progress of disease states.
ENZYME KINETICS We start with a very simple expression for formation of the enzyme-substrate complex, ES, from free enzyme, E, and free substrate, S. The rate constants for formation and breakdown back to reactants are k+1 and k-1, respectively. The rate constant for formation of the product is k+2, and to further simplify our treatment, we wil work within the approximation that we can neglect the backreaction, k-2. We can therefore write a simple kinetic scheme as follows: We will work within an approximation called the steady-state approximation, in which the rate of formation of the enzyme-substrate complex is equal to its rate of decay, and is therefore zero, i.e., It is also useful to write down two other definitions and assumptions before we continue: 1. The first thing we need to realize is that the total concentration of enzyme in all of its forms should be equal to the original free enzyme concentration: 2. The second point is that the rate of the reaction should be equal to formation of products, so the initial velocity, vo, and maximum possible velocity, or Vmax, obtained when all the enzyme is substrate-bound, can be written as Now, before making the steady-state approximation, we can look at the kinetic scheme above and deduce that the rate of change in the concentration of the enzyme-substrate complex should be equal to the rate of formation of the complex minus the rate of decay of the complex. The rate of formation is dependent upon the rate constant k+2 and the enzyme and substrate complexes. The rate of decay involves two possible pathways. One is to form products, and the other is to reform the reactants. Therefore the rate of decay of the enzyme-substrate complex will be the sum of two rate constants, k+2 and k-1. Therefore we can write and from the steady-state approximation, this has to equal zero. Before making this approximation, that equation is insoluble. Now, however, you can solve it fairly simply (despite what I did in class). First, if the steady-state approximation holds, then you can rearrange the equation to look like this: Now group all of the rate constants together We can define this collection of rate constants as something called the Michaelis Constant, or Km. Note that this is not an equilibrium constant. Now we have an even simpler expression:
We are almost done. However, we want an expression in terms of constants and measurable quantities such as the substrate concentration, [S], and the initial velocity, vo. To get the equation into that form, we need two things from above. The first is our statement that the total enzyme concentration is equal to the concentration of enzyme in all of its forms, which in turn is the same as the starting concentration of enzyme, i.e.,E total = Eo = [E] + [ES] or conveniently [E] = [Eo] [ES] more, . Then we can substitute that into our previously simplified equation to obtain Solve for ES Since initial velocity V0 = K2 [ES] and max velocity Vmax = K2 [E total]
Chapter-6 Factor affecting the rate of catalysis. 1234Substrate concentration. Temperature pH Inhibitors
The rate at which an enzyme works is influenced by several factors. 1. Concentration of substrate molecules (the more of them available, the quicker the enzyme molecules collide and bind with them). The concentration of substrate is designated [S] and is expressed in unit of molarity. 2. The temperature. As the temperature rises, molecular motion - and hence collisions between enzyme and substrate - speed up. But as enzymes are proteins, there is an upper limit beyond which the enzyme becomes denatured and ineffective. 3. The presence of inhibitors. The study of the rate at which an enzyme works is called enzyme kinetics. Many toxic substances owe their toxic properties to their ability to act as inhibitors (slow down the rate of reactions by different mechanism) to important enzymes responsible for catalyzing important biochemical processes. Once the enzyme is inhibited the process cannot take place, and a toxicological symptom occurs that often leads to paralysis, coma or even death of the organism. For example, cyanide poisoning is due to the cyanide ion competitively inhibiting the active site of the cytochromases enzymes responsible for catalyzing the Oxidation and Reduction processes of the Electron Transport System which is responsible for cellular respiration. Competitive Inhibition occurs when a molecule that is close enough to the shape of the true substrate will fit into the active site. Once locked into position, the blocker molecule prevents the true substrate molecule from getting into position. This effectively blocks the active site. The molecule competes for the active site with the true substrate molecule which is concentration dependent. If the concentration of substrate becomes more than the inhibitor they may replace the inhibitor later on. This is the reason why ethyl alcohol is given to person that has consumed the toxic alcohol (methyl alcohol) so that methyl alcohol can be competitively replaced by ethyl alcohol. Other inhibitors latch themselves not to the active site itself but to some portion of the enzyme molecule close to the active site which results in the
changing of the shape of the active site. This is referred to as non-competitive inhibition or mixed inhibition. Many heavy metals like Lead, Mercury, and Chromium will function as non-competitive inhibitors. Toxicology is the study of how toxicological substances can interfere with life sustaining enzymes via inhibition. The pesticide and herbicide industries make use of competitive and Non-Competitive Inhibitors. Biological warfare owes its success to enzyme inhibition but so does the life giving chemotherapeutic treatment of cancerous tumor growths with agents that inhibit important cancel cell enzymes. All in all the use of inhibitors can be used for the benefit of mankind or its destruction. 10.4 .1 Substrate concentration As we know that substrate react with the enzyme and it is converted into the product. If the substrate concentration is, low enough then the graph between the velocity of reaction and substrate utilization, will give almost a linear graph. But if the substrate concentration is high enough then we will obtain the graph that will be parabolic for some time and then straight in term of velocity. See the graph 10.9. FIGURE 10. 8 FIGURE 10. 9 10.4.2 Effect of temperature and pressure Every enzyme has a temperature range of optimum activity. Outside that temperature range the enzyme is rendered inactive and is said to be totally inhibited. This occurs because as the temperature changes this supplies enough energy to break some of the intramolecular attractions between polar groups (Hydrogen bonding, dipole-dipole attractions) as well as the Hydrophobic forces between non-polar groups within the protein structure. When these forces are disturbed and changed, this causes a change in the secondary and tertiary levels of protein structure, and the active site is altered in its conformation beyond its ability to accommodate the substrate molecules it was intended to catalyze. Most enzymes (and there are hundreds within the human organism) within the human cells will shut down at a body temperature below a certain value which varies according to each individual. This can happen if body temperature gets too low (hypothermia) or too high (hyperthermia). Temperature is an important factor in the regulation of enzyme activity. At some temperature activity becomes zero and at some temperature enzyme efficiency of conversion of substrate into product becomes high. Of all most important factor is the active site, because active site contains amino acid residues inside the pocket, and the ability of these residue to interact with the substrate functional group and capacity to breaking and making bonds is certainly going to be effected by the . Rates of all reactions, including those catalyzed by enzymes, rise with increase in temperature in accordance with the Arrhenius equation. K = A e G*/RT --------------27 OR log K = log A EA /2.3 RT where k is the kinetic rate constant for the reaction, A is the Arrhenius constant, also known as the frequency factor, G* =EA is the standard free energy of activation (kJ M-1) which depends on entropic and enthalpy factors, R is the gas law constant and T is the absolute temperature. Typical standard free energies of activation (15 - 70 kJ M-1) give rise to increases in rate by factors between 1.2 and 2.5 for every 10C rise in temperature. This factor for the increase in the rate of reaction for every 10C rise in temperature is commonly denoted by the term Q10 (i.e. in this case, Q10 is within the range 1.2 - 2.5). All the rate constants contributing to the catalytic mechanism will vary independently, causing
changes in both Km and Vmax. It follows that, in an exothermic reaction, the reverse reaction (having higher activation energy) increases more rapidly with temperature than the forward reaction. This, not only alters the equilibrium constant, but also reduces the optimum temperature for maximum conversion as the reaction progresses. The reverse holds for endothermic reactions such as that of glucose isomerase where the ratio of fructose to glucose, at equilibrium, increases from 1.00 at 55C to 1.17 at 80C. In general, it would be preferable to use enzymes at high temperatures in order to make use of this increased rate of reaction plus the protection it affords against microbial contamination. Enzymes, however, are proteins and undergo essentially irreversible denaturation (i.e.. conformational alteration entailing a loss of biological activity) at temperatures above those to which they are ordinarily exposed in their natural environment. These denaturing reactions have standard free energies of activation of about 200 - 300 kJ mole-1 (Q10 in the range 6 - 36) which means that, above a critical temperature, there is a rapid rate of loss of activity (Figure 8.5). The actual loss of activity is the product of this rate and the duration of incubation (Figure 8.6). It may be due to covalent changes such as the deamination of asparagine residues or non-covalent changes such as the rearrangement of the protein chain. Inactivation by heat denaturation has a profound effect on the enzymes productivity (Figure 8.7). ________________________________________ FIGURE 10. 10 FIGURE 10. 11 A schematic diagram showing the effect of the temperature on the activity of an enzyme catalyzed reaction. short incubation period; ----- long incubation period. Note that the temperature at which there appears to be maximum activity varies with the incubation time. ________________________________________ ________________________________________ FIGURE 10. 12 A schematic diagram showing the effect of the temperature on the productivity of an enzyme catalyzed reaction. 55C; 60C; 65C. The optimum productivity is seen to vary with the process time, which may be determined by other additional factors (e.g. overhead costs). It is often difficult to get precise control of the temperature of an enzyme catalyzed process and, under these circumstances, it may be seen that it is prudent to err on the low temperature side. The thermal denaturation of an enzyme may be modeled by the following serial deactivation scheme: --------27.1 Where kd1 and kd2 are the first-order deactivation rate coefficients, E is the native enzyme which may, or may not, be an equilibrium mixture of a number of species, distinct in structure or activity, and E1 and E2 are enzyme molecules of average specific activity relative to E of A1 and A2. A1 may be greater or less than unity (i.e. E1 may have higher or lower activity than E) whereas A2 is normally very small or zero. This model allows for the rare cases involving free enzyme (e.g. tyrosinase) and the somewhat commoner cases involving immobilised enzyme where there is a small initial activation or period of grace involving negligible discernible loss of activity during short incubation periods but prior to later deactivation. Assuming, at the beginning of the reaction: [E] = [E]0 -----------28 and: [E1] = [E2] = 0 ----29 At time t,
[E] = [E1] = [E2] = [E0] ----------30 It follows from the reaction scheme [27.1], - d[E] / dt =Kd1[E] -----------------31 Integrating equation 31 using the boundary condition in equation 28 gives: [E] = [E]0 e (-Kdt) = ---------------32 From the reaction scheme - d[E1] / dt =Kd2[E1]-Kd1 [E] ---------------33 Substituting for [E] from equation 32 - d[E1] / dt =Kd2[E1]-Kd1 [E]0 e (-Kdt) -------------34 Integrating equation 33 using the boundary condition in equation 29 gives: [E1]=Kd1 [E]0 / Kd2-Kd1 = (e (-Kd1t) - e (-Kd2t) ) -----------35 If the term 'fractional activity' (Af ) is introduced where, Af = [E] + A1 [E1] + A2 [E2] /[E0]---------36 then, substituting for [E2] from equation 30, gives: Af = [E] + A1 [E1] + A2 ([E0]-[E]-[E1]) /[E0]-------------37 therefore: ------38 When both A1 and A2 are zero, the simple first order deactivation rate expression results Af = e (-Kdt) -----------39 The half-life (t1/2) of an enzyme is the time it takes for the activity to reduce to a half of the original activity (i,e. Af = 0.5). If the enzyme inactivation obeys equation 39, the half-life may be simply derived, Ln (1/2) = =Kd1 t 1/2 -----------------------40 Therefore, t1/2 = 0.693 / Kd1 --------------41 In this simple case, the half-life of the enzyme is shown to be inversely proportional to the rate of denaturation. Many enzyme preparations, both free and immobilised, appear to follow this seriestype deactivation scheme. However because reliable and reproducible data is difficult to obtain, inactivation data should, in general, be assumed to be rather error-prone. It is not surprising that such data can be made to fit a model involving four determined parameters (A1, A2, kd1 and kd2). Despite this possible reservation, equations 38 and 39 remain quite useful and the theory possesses the definite advantage of simplicity. In some cases the series-type deactivation may be due to structural micro heterogeneity, where the enzyme preparation consists of a heterogeneous mixture of a large number of closely related structural forms. These may have been formed during the past history of the enzyme during preparation and storage due to a number of minor reactions, such as de amidation of one or two asparagine or glutamine residues, limited proteolysis or disulphide interchange. Alternatively it may be due to quaternary structure equilibrium or the presence of distinct genetic variants. In any case, the larger the variability the more apparent will be the series-type inactivation kinetics. The practical effect of this is that usually kd1 is apparently much larger than kd2 and A1 is less than unity. In order to minimise loss of activity on storage, even moderate temperatures should be avoided. Most enzymes are stable for months if refrigerated (0 - 4C). Cooling below 0C in the presence of additives (e.g. glycerol), which prevent freezing, can generally increase this storage stability even further. Freezing enzyme solutions is best avoided as it often causes denaturation due to the stress and pH variation caused by ice-crystal formation. The first order deactivation constants are often significantly lower in the case of enzyme-substrate, enzymeinhibitor and enzyme-product complexes which helps to explain the substantial stabilizing effects of suitable ligand, especially at concentrations where little free enzyme exists (e.g. [S] >> Km). Other factors, such as the presence of thiols anti-oxidants, may improve the thermal stability in particular cases. It has been found that the heat denaturation of enzymes is primarily due to the proteins' interactions with the aqueous environment. They are generally more
stable in concentrated, rather than dilute, solutions. In a dry or predominantly dehydrated state, they remain active for considerable periods even at temperatures above 100C. This property has great technological significance and is currently being exploited by the use of organic solvents. Pressure changes will also affect enzyme catalyzed reactions. Clearly any reaction involving dissolved gases (e.g. oxygenases and decarboxylases) will be particularly affected by the increased gas solubility at high pressures. The equilibrium position of the reaction will also be shifted due to any difference in molar volumes between the reactants and products. However an additional, if rather small, influence is due to the volume changes which occur during enzymic binding and catalysis. Some enzyme-reactant mixtures may undergo reductions in volume amounting to up to 50 ml mole-1 during reaction due to conformational restrictions and changes in their hydration. This, in turn, may lead to a doubling of the kcat, and/or a halving in the Km for a 1000 fold increase in pressure. The relative effects on kcat and Km depend upon the relative volume changes during binding and the formation of the reaction transition states. 10.4.3 Effect of pH on Enzyme Enzymes are amphoteric molecules containing a large number of acid and basic groups, mainly situated on their surface. The charges on these groups will vary, according to their acid dissociation constants, with the pH of their environment (Table 1.1). This will affect the total net charge of the enzymes and the distribution of charge on their exterior surfaces, in addition to the reactivity of the catalytically active groups. These effects are especially important in the neighborhood of the active sites. Taken together, the changes in charges with pH affect the activity, structural stability and solubility of the enzyme. Therefore all enzymes are affected by the change of pH. They act slowly on the low pH or high pH. So an optimum pH is needed for its full activity. Figure 10. 13 Figure showing optimum pH for two separate enzymes. This is because of change in the ionization pattern of the amino acid residue inside the active site of the enzyme. Either proton is removed or added. Changes in the pH or acidity of the environment can take place that would alter or totally inhibit the enzyme from catalyzing a reaction. This change in the pH will affect the polar and non-polar intramolecular attractive and repulsive forces and alter the shape of the enzyme and the active site as well to the point where the substrate molecule could no longer fit, and the chemical change would be inhibited from taking place as efficiently or not at all. In an acid solution any basic groups such as the Nitrogen groups in the protein would be protonated. If the environment was too basic the acid groups would be deprotonated. This would alter the electrical attractions between polar groups. Every enzyme has an optimum pH range outside of which the enzyme is inhibited. Some enzymes like many of the hydrolytic enzymes in the stomach such as Pepsin and Chymotrypsin effective operate at a very low acidic pH. Other enzymes like alpha amylase found in the saliva of the mouth operate most effectively at near neutrality. Still other enzymes like the lipases will function most effectively at basic pH values. If the pH drops in the blood called acidosis then enzymes in the blood will be inhibited outside their optimal pH range. If the pH climbs to an unacceptably high value called alkalosis then enzymes ceases to function effectively. Normally, these conditions do not take place because of the highly efficient buffers found in the blood that restrict the pH of the blood to a very narrow range. Buffers are a substance or mixtures of substances that resist any change in the pH. There are many buffer systems found in the body to adjust the pH so that enzymes might continue to catalyze their reactions. Correcting pH or temperature imbalances will usually allow the enzyme to resume its original shape or conformation. Some substances when added to the system will irreversibly break bonds disrupting the primary structure so that the enzyme is inhibited permanently. The enzyme is said to be irreversably denatured. Many toxic substances will break covalent bonds and cause the unraveling of the protein
enzyme. Other toxic substances will precipitate enzymes effectively removing them from the solution thus preventing them from catalyzing the reaction. This is also called denaturation. Table II pH for Optimum Activity Enzyme pH Optimum Lipase (pancreas) 8.0 Lipase (stomach) 4.0 - 5.0 Lipase (castor oil) 4.7 Pepsin 1.5 - 1.6 Trypsin 7.8 - 8.7 Urease 7.0 Invertase 4.5 Maltase 6.1 - 6.8 Amylase (pancreas) 6.7 - 7.0 Amylase (malt) 4.6 - 5.2 Catalase 7.0 There will be a pH, characteristic of each enzyme, at which the net charge on the molecule is zero. This is called the isoelectric point (pI), at which the enzyme generally has minimum solubility in aqueous solutions. In a similar manner to the effect on enzymes, the charge and charge distribution on the substrate(s), product(s) and coenzymes (where applicable) will also be affected by pH changes. Increasing hydrogen ion concentration will, additionally, increase the successful competition of hydrogen ions for any metal cationic binding sites on the enzyme, reducing the bound metal cation concentration. Decreasing hydrogen ion concentration, on the other hand, leads to increasing hydroxyl ion concentration which competes against the enzymes' ligand for divalent and trivalent cations causing their conversion to hydroxides and, at high hydroxyl concentrations, their complete removal from the enzyme. The temperature also has a marked effect on ionizations, the extent of which depends on the heats of ionization of the particular groups concerned. The relationship between the change in the pKa and the change in temperature is given by a derivative of the Gibbs-Helmholtz equation. where T is the absolute temperature (K), R is the gas law constant (8.314 J M-1 K1), H is the heat of ionisation and the numeric constant (2.303) is the natural logarithm of 10, as pKa's are based on logarithms with base 10. This variation is sufficient to shift the pI of enzymes by up to one unit towards lower pH on increasing the temperature by 50C. These charge variations, plus any consequent structural alterations, may be reflected in changes in the binding of the substrate, the catalytic efficiency and the amount of active enzyme. Both Vmax and Km will be affected due to the resultant modifications to the kinetic rate constants k+1, k-1 and kcat (k+2 in the Michaelis-Menten mechanism), and the variation in the concentration of active enzyme. The effect of pH on the Vmax of an enzyme catalysed reaction may be explained using the, generally true, assumption that only one charged form of the enzyme is optimally catalytic and therefore the maximum concentration of the enzyme-substrate intermediate cannot be greater than the concentration of this species. In simple terms, assume EH- is the only active form of the enzyme, The variation of activity with pH, within a range of 2-3 units each side of the pI, is normally a reversible process. Extremes of pH will, however, cause a timeand temperature-dependent, essentially irreversible, denaturation. In alkaline solution (pH > 8), there may be partial destruction of cystine residues due to base catalysed b-elimination reactions whereas, in acid solutions (pH < 4), hydrolysis of the labile peptide bonds, sometimes found next to aspartic acid residues, may occur. The importance of the knowledge concerning the variation of activity with pH cannot be over-emphasized. However, a number of other factors may mean that the optimum pH in the Vmax-pH diagram may not be the pH of choice in a
technological process involving enzymes. These include the variation of solubility of substrate(s) and product(s), changes in the position of equilibrium for a reaction, suppression of the ionization of a product to facilitate its partition and recovery into an organic solvent, and the reduction in susceptibility to oxidation or microbial contamination. The major such factor is the effect of pH on enzyme stability. This relationship is further complicated by the variation in the effect of the pH with both the duration of the process and the temperature or temperature-time profile. The important parameter derived from these influences is the productivity of the enzyme (i.e. how much substrate it is capable of converting to product). The variation of productivity with pH may be similar to that of the Vmax-pH relationship but changes in the substrate stream composition and contact time may also make some contribution. Generally, the variation must be determined under the industrial process conditions. It is possible to alter the pH-activity profiles of enzymes. The ionisation of the carboxylic acids involves the separation of the released groups of opposite charge. This process is encouraged within solutions of higher polarity and reduced by less polar solutions. Thus, reducing the dielectric constant of an aqueous solution by the addition of a co-solvent of low polarity (e.g. dioxan, ethanol), or by immobilisation, increases the pKa of carboxylic acid groups. This method is sometimes useful but not generally applicable to enzyme catalysed reactions as it may cause a drastic change on an enzyme's productivity due to denaturation (. The pKa of basic groups are not similarly affected as there is no separation of charges when basic groups ionise. However, protonated basic groups which are stabilised by neighbouring negatively charged groups will be stabilised (i.e. have lowered pKa) by solutions of lower polarity. Changes in the ionic strength (I) of the solution may also have some effect. The ionic strength is defined as half of the total sum of the concentration (ci) of every ionic species (i) in the solution times the square of its charge (zi); i.e. I=0.5(CiZi2) .For example, the ionic strength of a 0.1 M solution of CaCl2 , is 0.5 x (0.1 x 22 + 0.2 x 12) = 0.3 M. At higher solution ionic strength, charge separation is encouraged with a concomitant lowering of the carboxylic acid pKas. These changes, extensive as they may be, have little effect on the overall charge on the enzyme molecule at neutral pH and are, therefore, only likely to exert a small influence on the enzyme's isoelectric point. Chemical derivatisation methods are available for converting surface charges from positive to negative and vice-versa. It is found that a single change in charge has little effect on the pH-activity profile, unless it is at the active site. However if all lysines are converted to carboxylates (e.g. by reaction with succinic anhydride) or if all the carboxylates are converted to amines (e.g. by coupling to ethylene diamine by means of a carbodiimide the profile can be shifted about a pH unit towards higher or lower pH, respectively. The cause of these shifts is primarily the stabilisation or destabilisation of the charges at the active site during the reaction, and the effects are most noticeable at low ionic strength. The ionic strength of the solution is an important parameter affecting enzyme activity. This is especially noticeable where catalysis depends on the movement of charged molecules relative to each other. Thus both the binding of charged substrates to enzymes and the movement of charged groups within the catalytic 'active' site will be influenced by the ionic composition of the medium. If the charges are opposite then there is a decrease in the reaction rate with increasing ionic strength whereas if the charges are identical, an increase in the reaction rate will occur (e.g. the rate controlling step in the catalytic mechanism of chymotrypsin involves the approach of two positively charged groups, 57histidine+ and 145arginine+ causing a significant increase in kcat on increasing the ionic strength of the solution). Even if a more complex relationship between the rate constants and the ionic strength holds, it is clearly important to control the ionic strength of solutions in parallel with the control of pH.
FIGURE 10. 14 showing molecule activation at high and low temperature Enzyme Substrate Product Rate without Enzyme moles/L per min Rate with Enzyme moles/L per min Acceleration due to Enzyme Hexokinase Glucose Glucose 6-Phosphate <.0000001 1300 > 13 billion Phosphorylase <.000000005 1600 > 320 billion Alcohol Dehydrogenase Ethanol Acetaldehyde <.000006 2700 > 450 million Creatine Kinase Creatine Creatine Phosphate <.003 40 > 13, 000 o From: Daniel Koshland, Jr. Molecular geometry in enzyme action. Journal of Cellular & Comparative Physiology 47: 217-234, 1956.
Chapter-7 Enzyme inhibition A number of substances may cause a reduction in the rate of an enzyme catalysed reaction. Some of these (e.g. urea) are non-specific protein denaturants. Others, which generally act in a fairly specific manner, are known as inhibitors. Loss of activity may be either reversible, where activity may be restored by the removal of the inhibitor, or irreversible, where the loss of activity is time dependent and cannot be recovered during the timescale of interest. If the inhibited enzyme is totally inactive, irreversible inhibition behaves as a time-dependent loss of enzyme concentration (i.e.. lower Vmax), in other cases, involving incomplete inactivation.There may be time-dependent changes in both Km and Vmax. Heavy metal ions (e.g. mercury and lead) should generally be prevented from coming into contact with enzymes as they usually cause such irreversible inhibition by binding strongly to the amino acid backbone. More important for most enzyme-catalysed processes is the effect of reversible inhibitors. These are generally discussed in terms of a simple extension to the Michaelis-Menten reaction scheme. --------------------42 where I represents the reversible inhibitor and the inhibitory (dissociation) constants Ki and Ki' are given by --------------------43 and, --------------------------44 For the present purposes, it is assumed that neither EI nor ESI may react to form product. Equilibrium between EI and ESI is allowed, but makes no net contribution to the rate equation as it must be equivalent to the equilibrium established through: ------45 Binding of inhibitors may change with the pH of the solution, as discussed earlier for substrate binding, and result in the independent variation of both Ki and Ki' with pH.
In order to simplify the analysis substantially, it is necessary that the rate of product formation (k+2) is slow relative to the establishment of the equilibria between the species. Therefore: -----------46 also ----------------------------47 where: -----------48 therefore ---------------49 Substituting from equations (43), ( 44) and (46), followed by simplification, gives: --------------50 therefore --------------51 If the total enzyme concentration is much less than the total inhibitor concentration (i.e. [E]0<< [I]0), then: --------------------52 This is the equation used generally for mixed inhibition involving both EI and ESI complexes (Figure 10.19a). A number of simplified cases exist that are reversible. 1. Competitive inhibition. 2. Noncompetitive inhibition 3. Uncompetitive inhibition. Figure 11. 1 showing effect of concentration of inhibitor on enzyme activity on different time interval. A. Competitive inhibition This occurs when both the substrate and inhibitor compete for binding to the active site of the enzyme. The inhibition is most noticeable at low substrate concentrations but can be overcome at sufficiently high substrate concentrations as the Vmax remains unaffected (Figure 10.19b). Normal Vmax can be observed when substrate is sufficiently present. In the presence of a competitive inhibitor, it takes a higher substrate concentration to achieve the same velocities that were reached in its absence. So while Vmax can still be reached if sufficient substrate is available, one-half Vmax requires a higher [S] than before and thus Km is larger. But at [S] at which Vo=1/Vmax, the Km (apparent Km) will increase in presence of inhinitor. In double reciprocal plot this can be obsereved. Ki' is much greater than the total inhibitor concentration and the ESI complex is not formed but EI is formed. The rate equation is given by: ---------------------52 where Kmapp is the apparent Km for the reaction, and is given by -----------------53 Normally the competitive inhibitor bears some structural similarity to the substrate, and often is a reaction product (product inhibition, e.g. inhibition of lactase by galactose), which may cause a substantial loss of productivity when high degrees of conversion are required. The rate equation for product inhibition is derived from equations (52) and (53). -----54 Figure 11. 2 figure showing competitive inhibition 1/Vo (1/ M/min) verses 1/[S] (1/ mM). note that on increasing inhibitor concentration 1/Vmax remain unchanged while Km value changes. Figure 11. 3 showing effect of concentration of inhibitor on enzyme activity.
A similar effect is observed with competing substrates, quite a common state of
affairs in industrial conversions, and especially relevant to macromolecular hydrolyses where a number of different substrates may coexist, all with different kinetic parameters. The reaction involving two co-substrates may be modelled by the scheme. ---------------55 Both substrates compete for the same catalytic site and, therefore, their binding is mutually exclusive and they behave as competitive inhibitors of each others reactions. If the rates of product formation are much slower than attainment of the equilibria (i.e. k+2 and k+4 are very much less than k-1 and k-3 respectively), the rate of formation of P1 is given by: ---------56 and the rate of formation of P2 is given by ---------------57 If the substrate concentrations are both small relative to their Km values: --------------------58 Therefore, in a competitive situation using the same enzyme and with both substrates at the same concentration: --------------59 Where and > in this simplified case. The relative rates of reaction are in the ratio of their specificity constants. If both reactions produce the same product (e.g. some hydrolyses): ---------------60 therefore --------------------------61 B. Uncompetitive inhibition This occurs when the inhibitor binds to a site which only becomes available after the substrate (S1) has bound to the active site of the enzyme. Ki is much greater than the total inhibitor concentration and the EI complex is not formed. This inhibition is most commonly encountered in multi-substrate reactions where the inhibitor is competitive with respect to one substrate (e.g. S2) but uncompetitive with respect to another (e.g. S1), where the reaction scheme may be represented by. --------61 The inhibition is most noticeable at high substrate concentrations (i.e. S1 in the scheme above) and cannot be overcome as both the Vmax and Km are equally reduced (Figure 1.8c). The rate equation is: ----------------------62 where Vmaxapp and Kmapp are the apparent Vmax and Km given by --------------------63 and --------------------64 Figure 11. 4
Figure 11. 5 figure showing case of uncompetitive inhibition In this case the specificity constant remains unaffected by the inhibition. Normally the uncompetitive inhibitor also bears some structural similarity to one of the substrates and, again, is often a reaction product. ________________________________________ figure 11.6 A schematic diagram showing the effect of reversible inhibitors on the rate of enzyme-catalysed reactions. no inhibition, (a) mixed inhibition ([I] = Ki = 0.5 Ki'); lower Vmaxapp (= 0.67 Vmax), higher Kmapp (= 2 Km). (b) competitive inhibition ([I] = Ki); Vmaxapp unchanged (= Vmax), higher Kmapp (= 2 Km). (c) uncompetitive inhibition ([I] = Ki'); lower Vmaxapp (= 0.5 Vmax) and Kmapp (= 0.5 Km). (d) noncompetitive inhibition ([I] = Ki = Ki'); lower Vmaxapp (= 0.5 Vmax), unchanged Kmapp (= Km). ________________________________________
A special case of uncompetitive inhibition is substrate inhibition which occurs at high substrate concentrations in about 20% of all known enzymes (e.g. invertase is inhibited by sucrose). It is primarily caused by more than one substrate molecule binding to an active site meant for just one, often by different parts of the substrate molecules binding to different sites within the substrate binding site. If the resultant complex is inactive this type of inhibition causes a reduction in the rate of reaction, at high substrate concentrations. It may be modeled by the following scheme ---------------65 where ---------------------66 The assumption is made that ESS may not react to form product. It follows from equation (52) that: -------------------------67 Even quite high values for KS lead to a leveling off of the rate of reaction at high substrate concentrations, and lower KS values cause substantial inhibition (Figure 10.20). ________________________________________ Figure 11. 7 The effect of substrate inhibition on the rate of an enzymecatalysed reaction. A comparison is made between the inhibition caused by increasing KS relative to Km. no inhibition, KS/Km >> 100; KS/Km = 100; KS/Km = 10; KS/Km = 1. By the nature of the binding causing this inhibition, it is unlikely that KS/Km < 1. ________________________________________ C. Noncompetitive inhibition This occurs when the inhibitor binds at a site away from the substrate binding site, causing a reduction in the catalytic rate. The inhibitor binding causes inactivation of enzyme due to which apparent Vmax (Vmax = Kcat [Et] ) lower down whether or not substrate is bound. enzyme rate (velocity) is reduced for all values of [S], including Vmax and one-half Vmax but Km remains unchanged because the active site of those enzyme molecules that have not been inhibited is unchanged Therefore there is no effect on Km. It is quite rarely found as a special case of mixed inhibition. Both the EI and ESI complexes are formed equally well (i.e. Ki equals Ki'). The fractional inhibition is identical at all substrate concentrations and cannot be overcome by increasing substrate concentration due to the reduction in Vmax . The rate equation is given by: Figure 11. 8 figure showing Noncompetative inhibition slope is Km /Vmax ---------------------------68 where Vmaxapp is given by: --------------------------------69 The diminution in the rate of reaction with pH, described earlier, may be considered as a special case of noncompetitive inhibition; the inhibitor being the hydrogen ion on the acid side of the optimum or the hydroxide ion on the alkaline side. Figure 11. 9
Figure 11. 6 (b) Determination of Vmax and Km It is important to have as thorough knowledge as is possible of the performance characteristics of enzymes, if they are to be used most efficiently. The kinetic parameters Vmax, Km and kcat/Km should, therefore, be determined. There are two approaches to this problem using either the reaction progress curve (integral
method) or the initial rates of reaction (differential method). Use of either method depends on prior knowledge of the mechanism for the reaction and, at least approximately, the optimum conditions for the reaction. If the mechanism is known and complex then the data must be reconciled to the appropriate model (hypothesis), usually by use of a computer-aided analysis involving a weighted least-squares fit. Many such computer programs are currently available and, if not, the programming skill involved is usually fairly low. If the mechanism is not known, initial attempts are usually made to fit the data to the Michaelis-Menten kinetic model. ------------------70 which, on integration, using the boundary condition that the product is absent at time zero and by substituting [S] by ([S]0 - [P]), becomes ------------71 If the fractional conversion (X) is introduced, where -----------------------72 then equation (72) may be simplified to give: ----------73 Use of equation (70) involves the determination of the initial rate of reaction over a wide range of substrate concentrations. The initial rates are used, so that [S] = [S]0, the predetermined and accurately known substrate concentration at the start of the reaction. Its use also ensures that there is no effect of reaction reversibility or product inhibition which may affect the integral method based on equation (73). Equation (70) can be utilised directly using a computer program, involving a weighted least-squares fit, where the parameters for determining the hyperbolic relationship between the initial rate of reaction and initial substrate concentration (i.e.. Km and Vmax) are chosen in order to minimise the errors between the data and the model, and the assumption is made that the errors inherent in the practically determined data are normally distributed about their mean (error-free) value. Alternatively the direct linear plot may be used (Figure 10.25). This is a powerful non-parametric statistical method which depends upon the assumption that any errors in the experimentally derived data are as likely to be positive (i.e. too high) as negative (i.e. too low). It is common practice to show the data obtained by the above statistical methods on one of three linearised plots, derived from equation (70) (Figure 10.25). Of these, the double reciprocal plot is preferred to test for the qualitative correctness of a proposed mechanism, and the Eadie-Hofstee plot is preferred for discovering deviations from linearity. ________________________________________ Figure 11. 7 The direct linear plot. A plot of the initial rate of reaction against the initial substrate concentration also showing the way estimates can be directly made of the Km and Vmax. Every pair of data points may be utilised to give a separate estimate of these parameters (i.e. n(n-1)/2 estimates from n data points with differing [S]0). These estimates are determined from the intersections of lines passing through the (x,y) points (-[S]0,0) and (0,v); each intersection forming a separate estimate of Km and Vmax. The intersections are separately ranked in order of increasing value of both Km and Vmax and the median values taken as the best estimates for these parameters. The error in these estimates can be simply determined from sub-ranges of these estimates, the width of the subrange dependent on the accuracy required for the error and the number of data points in the analysis. In this example there are 7 data points and, therefore, 21 estimates for both Km and Vmax. The ranked list of the estimates for Km (mM) is 0.98,1.65, 1.68, 1.70, 1.85, 1.87, 1.89, 1.91, 1.94, 1.96, 1.98, 1.99, 2.03, 2.06, 1.12, 2.16, 2.21, 2.25, 2.38, 2.40, 2.81, with a median value of 1.98 mM. The Km must lie between the 4th (1.70 mM) and 18th (2.25 mM) estimate at a confidence level of 97% (Cornish-Bowden et al., 1978). The list of the estimates for Vmax ( M.min-1) is ranked separately as 3.45, 3.59, 3.80, 3.85, 3.87, 3.89, 3.91, 3.94, 3.96, 3.96, 3.98, 4.01, 4.03, 4.05, 4.13, 4.14, 4.18, 4.26, 4.29, 4.35, with a
median value of 3.98 M.min-1. The Vmax must lie between the 4th (3.85 M.min-1) and 18th (4.18 M.min-1) estimate at a confidence level of 97%. It can be seen that outlying estimates have little or no influence on the results. This is a major advantage over the least-squared statistical procedures where rogue data points cause heavily biased effects. ________________________________________ Three ways in which the hyperbolic relationship between the initial rate of reaction and the initial substrate concentration can be rearranged to give linear plots (a) Lineweaver-Burk (double-reciprocal) plot of 1/v against 1/[S]0 giving intercepts at 1/Vmax and -1/Km --------------74 Figure 11. 8 (a) Lineweaver-Burk (double-reciprocal) plot of 1/v against 1/[S]0 giving intercepts at 1/Vmax and -1/Km Figure 11. 9 (b) Eadie-Hofstee plot of v against v/[S]0 giving intercepts at Vmax and Vmax/Km (75) Figure 11. 10 c Hanes-Woolf (half-reciprocal) plot of [S]0/v against [S]0 giving intercepts at Km/Vmax and Km. ----------76 The progress curve of the reaction (Figure 10.28) can be used to determine the specificity constant (kcat/Km) by making use of the relationship between time of reaction and fractional conversion (see equation (73). This has the advantage over the use of the initial rates (above) in that fewer determinations need to be made, possibly only one progress curve is necessary, and sometimes the initial rate of reaction is rather difficult to determine due to its rapid decline. If only the early part of the progress curve, or its derivative, is utilised in the analysis, this procedure may even be used in cases where there is competitive inhibition by the product, or where the reaction is reversible. ________________________________________ Figure 11. 11 FIGURE 10. 15 . A schematic plot showing the amount of product formed (productivity) against the time of reaction, in a closed system. The specificity constant may be determined by a weighted least-squared fit of the data to the relationship given by equation (73). Table -2
Module III Enzyme Kinetics: Single substrate steady state kinetics; Michaelis Menten equation, Linear plots, King-Altmans method; Inhibitors and activators; Multisubstrate systems; ping-pong mechanism, Alberty equation, Sigmoidal kinetics and Allosteric enzymes Chapter-8 Multisubstrate enzymes Many Enzymes react with two or more substrates simultaneously (includes cofactors : ATP, NADPH, NADP, FADH, FAD etc) Michaelis Menten kinetics is observed when only one substrate is varied with the other(s) held constant (usually the cofactors) Common observed Mechanisms: Sequential All substrates are bound by enzyme before the any products are formed and released Ordered : Substrates bind and products released in specific order Random : no order Ping Pong When one or more product(s) are released before all substrates are bound by enzyme (acyl/phosphoryl enzyme intermediate) sequential means all substrates on before any products off, while ping pong has products off before all substrates on - can be ordered or random - e.g.: ________________________________________ Adenylate kinase displays sequential kinetics, in which both substrates must be bound before any product is released. This is distinguished from what is termed ping-pong kinetics, in which one reactant modifies the enzyme, and then a second reactant interacts with the modification. Sequential kinetics can be distinguished from ping-pong kinetics by initial rate studies. In practice, measure initial rates as a function of one substrate while holding the other constant. Then, vary the concentration of the second substrate and repeat. Lineweaver-Burk (double-reciprocal) analysis should yield a family of lines that intersect at the left of the y-axis of the graph. Within the realm of sequential reactions lies ordered sequential and random sequential at the extreme ends. In ordered sequential reactions, one substrate is obligated to bind to the enzyme before a second substrate. In random sequential mechanisms there is no preference. In practice, there is usually some degree of order in binding.
DG= refers to the difference in free energy between the transition state and the substrate Enzymes work by decreasing DG= to facilitate more rapid formation of P Think of it as enzymes helping the chemical reaction get over the hump in order to form product (P) ES complex demonstrated in a variety of ways including X-ray crystallography, electron microscopy, and spectrophotometry Enzymes derive much of their catalytic power by bringing in a substrate molecule at a favorable orientation (this is how enzymes reduce the free energy required to convert S into P) Leonor Michaelis (1913): noticed that at constant [enzyme] the rate of a reaction increases with increasing [S] until a maximal velocity (Vmax) is achieved This saturation effect is an important distinction versus uncatalyzed reactions Interpretation of data: ES complexes formed until substrate saturation occurs at which point no more substrate binding sites (i.e., enzyme molecules) are available The kinetic mechanism of an enzyme is simply the sequence in which substrates bind to, and products are released from the enzyme. It should not be confused with the chemical mechanism which is concerned with the chemical interactions between the enzyme and its substrate(s) which results in the creation of product(s). Kinetic mechanisms can be broadly divided into two main types, sequential and ping-pong (also known as double displacement) mechanisms. Sequential mechanisms Sequential reactions are ones in which all reactants bind to the enzyme before the first product is released. They can be further subdivided into ordered reactions, in which the reactants and products are bound and released in an obligatory sequence, and random reactions, in which there is no obligatory binding sequence. Ping-pong mechanisms Ping-pong reactions are ones in which at least one product is released before all the substrates have bound. To clarify these distinctions we'll look at each of these mechanisms in turn using a typical bi bi enzyme: A + B P + Q We'll start by looking at an ordered sequential reaction, which is perhaps the simplest in kinetic terms. Types of specificity: o Geometric specificity: shape Chiral specificity: most chirally specific enzymes are absolutely stereospecific. Prochirality, because of their own chiral nature enzymes can often hold substrates in such a way that on one chiral product is made, distinguishing between seemingly identical groups. o Chemical specificity: functional groups, types of chemical reaction. Enzyme Catalysis We will look at catalysis in two types of systems: Model systems in organic chemistry to elucidate probable mechanisms of chemical catalysis. Example enzymes to demonstrate these mechanisms in enzyme catalysis.
Mechanisms of Chemical Catalysis Look at some examples of catalysis in model systems (organic chemistry) and how they might operate in enzymes. Types of Catalysis: Acid/Base o Specific (H+ & OH- in water) o General (Bronsted acid definition: proton donor/acceptor pairs; buffers.) Covalent o nucleophilic o electrophilic Proximity/orientation Stabilization of Transition State Conformation (Strain/distortion; Charge neutralization) Metal/Metal ion. So how does catalysis work? Recall that the slow step of a reaction is reaching the transition state. Thus if we can find a way to stabilize the transition state (lower Ea) then the reaction rate will be enhanced. Generally we will be looking at three ways to increase rates 1. stabilize transition states 2. increase the concentrations of intermediates 3. use a different reaction pathway. o Plots of vi = d[P]/dt vs. [S] for 0 - 3rd order First Order: r = k[S] for S P; & r =-d[S]/dt = d[P]/dt. o Example - SN1 from organic chemistry: A + B C + D as 1st order reaction, as in the hydroxylation of t-alkylchloride: First order because rate depends only on the formation of the carbocation, which in turn depends only on [R3CCl] Second order: r = k[A][B] for A + B P; or can have r = k[A]2 ; o Example - SN2 from organic chemistry: A + B C + D as 2nd order reaction, as in the hydroxylation of primary alkylchlorides: Now 2nd order because both RH2CCl and OH- (reverse of figure above) are involved in slow step. Higher order reactions occur, but uncommon. Zero order: r = k: Only occurs with catalysts, important in enzyme catalysis. 0 order also only occurs above a minimum [A]. one-substrate enzymes: For simple enzyme, S P get rectangular hyperbola type plot for vi vs [S] (text Figure 6-11), similar to Mb binding curve. Let's look at a mathematical model and attempt to generate curve. This was first done by Michaelis and Menten for an equilibrium model. Better is the steady state model of Haldane and Briggs (more general), which we will derive. For S P assume And for initial reaction conditions [P] = 0 & therefore k4 = 0, so have Now vi = d[P]/dt = k3[ES] (Note that kcat is often used instead of k3); Assume steady state (steady state assumption: d[ES]/dt= 0): d[ES]/dt= 0; Thus: 0 = d[ES]/dt= k1[E][S] - k2[ES] - k3[ES]. So, Which is known as the Michaelis-Menten Equation. For simple, one-substrate enzymes then, have Michaelis-Menten Equation as a model for enzyme activity.
Note predicted consequences of model: [S] >> KM; then vi = Vmax and get Zero order (r = k) [S] << KM; then , and get First order (r = k [S]) [S] = KM; then vi = Vmax/2 This is definition of KM, the substrate concentration at half-saturation. Note consequences for a plot: start off with approximately linear slope with y = kx. Then at the limit of high concentrations have a horizontal line. This is exactly what we expect if we look at the general form of the equation: , the formula for a rectangular hyperbola: (text Figure 6-12) Turnover Number. The rate constant (First order) for the breakdown of the [ES] complex, kcat (k3), is also known as the turnover number, that is the maximum number of substrate molecules processed/active site (moles substrate/mole active site): kcat=Vmax / [E]total. Note that this is best determined under saturating conditions. (text Table 6-08) At very low concentrations of [S] can find the second-order rate constant for the conversion of E + S E + P: vo = (kcat / KM)([E][S]. Linear plots for enzyme kinetic studies Double Reciprocal or Lineweaver-Burke Plot: Need in form: y = ax + b , so take reciprocals of both sides and have . (text Box 6-01 figure 6-1) Other linear plots are also available, and are better in terms of statistics (L-B one of worst, best quality points [high concentration] have least influence on slope, while low precision points [low concentration] are more spread out, and have a large moment, with a strong influence on the slope and the KM intercept this is not as much of a concern now with computer statistical packages, but you still have to understand the statistics). We will see others in the laboratory discussion. FYI - The Eadie-Hofstee Plot One common plot is shown below. Note that the data points are distributed much more evenly over the plot giving better statistics for the slope. In addition the value of KM is obtained from the slope, giving better precision.
ENZYME KINETICS AND INHIBITION What's exciting about enzyme inhibition? Potential to tell us about enzyme. Potential uses as drugs and toxins. o Understanding drugs and toxins to counter etc. Three major types of inhibition: Competitive inhibition Noncompetitive inhibition Uncompetitive inhibition Competitive Inhibition: S & I are mutually exclusive, E can bind to one OR the other. Plots: (text Box 6-2 figure 1) We can model this inhibition with chemical equations, keeping in mind that S & I are mutually exclusive, E can bind to one OR the other: (text Figure 6-15a) Model: ; and: ; where . Classically assume binding to same site, but other possibilities also. 1. steric hindrance between S & I in different sites. 2. overlapping sites for S & I. 3. Partial sharing of sites.
4. Conformational change of enzyme with binding of either such that other can not bind. Noncompetitive: the inhibitor can bind to either E or ES. S & I do not bind to the same sites! (text Figure 6-15c) Model: ; and . Note that will have two inhibitor binding constants, they may be the same, as in the equation above, or could be different, leading to more complex behavior. Plots for classic, simple situation (overhead MvH 11.5): Mixed: Like non-competitive above, Model: ; and but with different values of Ki. This is in fact the more common situation, with a L-B plot with an intersection on the plot above the 1/vo axis. (text Box 6-2 figure 3) Uncompetitive: In uncompetitive inhibition the inhibitor binds ONLY to the ES complex (text Figure 6-15b). Model: ; and . For double reciprocal plots get parallel lines! (text Box 6-2 figure 2) This is not generally found for single substrate enzymes, but is found in multi-substrate systems. Multi-substrate Enzymes Look at three common and easily understood types. We will use Cleland Nomenclature and "Kinetic mechanism diagrams." Ordered Sequential Bi Bi mechanism (two on; two off); Note: A must bind first, Q is released last. Ping Pong Bi Bi (one on, one off; one on, one off); Note: have some sort of modified enzyme intermediate (often covalent intermediate) Random Sequential Bi Bi (two on; two off); Note: A or B may bind first, P or Q may be released last. Two-substrate Enzyme Product Inhibition Patterns (Based on: E. B. Cunningham, Biochemistry: Mechanisms of Metabolism. McGraw-Hill Book Company, New York (1978), and W. Cleland, "Substrate Inhibition: in Contemporary Enzyme Kinetics and Mechanism. (Daniel L. Purich, ed.) Academic Press, New york (1983)) Kinetic Mechanism Variable Substrate Product Type of Inhibition Ordered Sequential Bi Bi Ordered Sequential Bi Bi A B Q Noncompetitive A P Noncompetitive B P Noncompetitive Random Sequential Random Sequential B Q A P B P Ping pong Bi Bi Ping pong Bi Bi B Q A P B P Bi Bi Bi Bi A Q Noncompetitive Noncompetitive Noncompetitive Q Competitive
Noncompetitive
A Q Competitive Noncompetitive Noncompetitive Competitive
Note that in each case we can predict/explain the pattern of inhibition on the
basis of the substrate and inhibitor binding to the same "enzyme form." Thus for the Ordered Sequential mechanism only the first substrate and last product bind to the same form, in this case the free enzyme. Similarly for the Ping pong mechanism the first substrate and last product should be competitive as the both bind the free enzyme. In this case we also see a competitive inhibition between the second substrate and the first product, since they both bind to the E-X complex. The Random Sequential mechanism is a bit more subtle. Here we see across the board noncompetitive since in each case the substrates (and products) can each bind to more than one substrate form, so competitive inhibition will not be possible! (Think of the product as competing with one order of binding but not the other.) The ping-pong (double displacement) mechanism ________________________________________ The distinguishing feature of these enzymes is that at least one product is released from the enzyme before all of the substrates have bound. This might seem slightly unlikely at a first glance, but it's actually easily explained and quite a common mechanism. Some very familiar enzymes, for instance the serine proteases (trypsin, chymotrypsin, etc.) and the amino transferases work in this way. The process starts by binding of the enzyme to the first substrate in the usual way: E + A (EA) Notice that I've used parentheses around the EA complex to indicate that it is a central complex. The active site is full as this substrate will be converted to product before the second substrate can bind. The active site has no room for the second substrate so it is full. The next reaction is the key to the whole process: (EA) (FP) In this reaction a part of the substrate has been removed from substrate A, converting it to product P. The removed section has become covalently bound to the enzyme to create a new form of the enzyme, enzyme F. The first product of the reaction is now released and the second substrate binds: (FP) F + P F + B (FB) Now the stored section of the first substrate is transferred to the second substrate to create the second product, which is then released: (FB) (EQ) (EQ) E + Q The animation should help to clarify this. The Cleland plot for a ping-pong enzyme is quite distinctive: as the first upward arrow (product P) is to the left of the first down arrow (substrate B). Now that we're familiar with the principal types of kinetic mechanism we need to think about experimental techniques to distinguish between them. To start with we'll examine the effects of changes in substrate concentration on multisubstrate reactions. Effects of Substrate Concentration in Multisubstrate Systems ________________________________________ With a multisubstrate enzyme, let's say a typical bi bi enzyme: A + B P + Q we can carry out exactly the same experiment. We simply need to keep one substrate (the fixed substrate) at a constant concentration in all our assays, while we vary the concentration of the other substrate (the variable substrate). The results would be exactly the same as you'd expect in a single substrate system and you could use any of the methods that we've studied to calculate the kinetic parameters. What would happen though if you repeated the experiment with an increased concentration of the fixed substrate. Since you're increasing the concentration of a substrate you would expect the velocity to rise and in fact the
reaction would be faster at any given concentration of the variable substrate than it was in the previous experiment. So you'd end up with a second set of data which you could use in your chosen plot. The kinetic parameters would change to reflect the change in velocity. If you repeated this at a variety of concentrations of the fixed substrate you would get a series of lines. A typical Lineweaver-Burk plot obtained as a result of this type of experiment can be seen below. Substrate A was used as the variable substrate and substrate B as the fixed substrate. The actual pattern of lines obtained will vary according to the way in which the enzyme interacts with the two substrates and, as we'll see in the next couple of pages, enables us to distinguish between sequential and ping-pong enzymes. In discussing graphs of this type we'll be considering changes in the Vmax and slope of the line. A change in Vmax indicates the effect that a change in the concentration of the fixed substrate on the reaction speed at very high concentrations of the variable substrate. Remember that Vmax is the velocity of the reaction under those conditions. A change in slope indicates the effect of a change in concentration of the fixed substrate on the speed at very low concentrations of the variable substrate. Remember in our discussion of enzyme inhibition we found that the slope was the rate constant at low substrate concentrations. We'll start by considering the expected results of this experiment when carried out with a sequential enzyme. Substrate concentration assays with sequential enzymes ________________________________________ Consider a bi bi ordered sequential reaction: A + B P + Q The Cleland plot for such a reaction would be: We'll discuss the results of a set of enzyme assays in which substrate A is used as the variable substrate and substrate B as the fixed substrate. At very low A concentrations the rate limiting step of the reaction would be the binding of A to the enzyme as the substrate is in very short supply. An increase in the concentration of B would reduce the concentration of the EA complex in the reaction mixture by binding to it to form EAB complex. Reducing the concentration of the product of the E + A EA reaction will pull it to the right by the Law of Mass Action. So the increase in B has increased the speed of the rate limiting step, and therefore of the overall reaction. As we're looking at effects at low A concentrations this would be seen as a change in the slope of the Lineweaver-Burk plot. At very high A concentrations the rate limiting step would be the EAB EPQ, which is the inherently slowest reaction, or EA + B EAB at low B levels. An increase in B would increase the speed of either of these as a reactant in the second reaction and by the knock on effect of generating more EAB for the central reacton to occur. This would be seen as a change in Vmax as we are considering effects at high A concentration. In summary then, for an ordered sequential reaction, a change in the concentration of the fixed substrate would bring about a change in both the slope and intercept of the Lineweaver-Burk plot and the graph would be similar to that suggested on the previous page. A similar result would be obtained if the assay was reversed and B used as the variable substrate or if the enzyme used a random sequential mechanism. You might like to demonstrate that yourself as an exercise.
Chapter-9 ISOLATION AND PURIFICATION OF ENZYME INTRODUCTION ENZYME is isolated and purified to obtain maximum desired product because they are not utilized during the product formation. And also isolation of enzyme gives the maximum understanding of the behavior of the enzyme in complex system, its regulation mechanism. PURIFIED enzyme is needed for laboratory work and less purified enzyme is needed for commercial purpose. Purifying enzyme is much labor intensive process and main aim is to separate other protein that are not the part of the enzyme. Often during purification enzyme yield is decreases many folds. Purification of enzyme is aimed to get maximum turn over number that is to increase the activity of enzyme in other world to increase its efficiency of conversion of per mole substrate into the product. It helps in increasing the sensitivity of the diagnosis test in the clinics. Other need of the enzyme purification is the study of kinetics that is rate of enzyme and its specificity towards the substrate. Inhibition kinetics helps in functional study of the enzyme. During the disturbance in the metabolic pathway some enzyme may not form and it results in the formation of unwanted product like in the phenylketonuria disease homigenistic acid secretion in the urine results. Therefore for the diagnostic purpose much pure enzyme is needed. FIGURE 8. 1 strategies for enzyme purification. source of enzyme Method of enzyme isolation (cell lysis by osmotic, enzyme, sonication, or homogenization method) Method of separation. [i] Based on size and mass ( centrifugation, GPC gel permeation chromatography, Dialysis and ultracentrifugation). [ii] Based on polarity (Ion exchange, electrophoresis, isoelectric focusing, hydrophobic interaction chromatography). [iii] Based on changes in solubility ( by change of pH, change in ionic strength (Salting in or salting out). [iv] Based on change in dielectric strength by adding organic solvent. [v] Based on specific binding site. Affinity chromatography. Affinity elution. Dye ligand chromatography. Immuno-adsorption chromatography. Covalent chromatography. (4). Test of purification a). test of purity b). Test of catalytic activity c). Active site titration Test of purity is done by following method 1. Ultrapurification (for impurities < 5%) 2. Electrophoresis (Examining enzyme composed of non identical subunit) 3. SDS-PAGE ( good method for detecting impurities and for detecting damage of non-identical subunit.) 4. Capillary Electrophoresis 5. Isoelectric focusing ( very sensitive method) 6. Mass spectrometry.( power full method ) Source of enzyme
Biologically active enzymes may be extracted from any living organism. A very wide range of sources are used for commercial enzyme production from Actinoplanes to Zymomonas, from spinach to snake venom. Of the hundred or so enzymes being used industrially, over a half are from fungi and yeast and over a third are from bacteria with the remainder divided between animal (8%) and plant (4%) sources (Table 8.1). A very much larger number of enzymes find use in chemical analysis and clinical diagnosis. Non-microbial sources provide a larger proportion of these, at the present time. Microbes are preferred to plants and animals as sources of enzymes because: 1. they are generally cheaper to produce. 2. their enzyme contents are more predictable and controllable, 3. reliable supplies of raw material of constant composition are more easily arranged, and 4. plant and animal tissues contain more potentially harmful materials than microbes, including phenolic compounds (from plants), endogenous enzyme inhibitors and proteases. 5. Attempts are being made to overcome some of these difficulties by the use of animal and plant cell culture. ________________________________________ Table 2.1. Some important industrial enzymes and their sources. Enzyme a EC number b Source Intra/extra -cellular c Scale of production d Industrial use Animal enzymes Catalase 1.11.1.6 Liver I Food Chymotrypsin 3.4.21.1 Pancreas E Leather Lipasee 3.1.1.3 Pancreas E Food Rennetf 3.4.23.4 Abomasum E + Cheese Trypsin 3.4.21.4 Pancreas E Leather Plant enzymes Actinidin 3.4.22.14 Kiwi fruit E Food Amylase 3.2.1.1 Malted barley E +++ Brewing -Amylase 3.2.1.2 Malted barley E +++ Brewing Bromelain 3.4.22.4 Pineapple latex E Brewing Glucanaseg 3.2.1.6 Malted barley E ++ Brewing Ficin 3.4.22.3 Fig latex E Food Lipoxygenase 1.13.11.12 Soybeans I Food Papain 3.4.22.2 Pawpaw latex E ++ Meat Bacterial enzymes Amylase 3.2.1.1 Bacillus E +++ Starch -Amylase 3.2.1.2 Bacillus E + Starch Asparaginase 3.5.1.1 Escherichia coli I Health Glucose isomeraseh 5.3.1.5 Bacillus I ++ Fructose syrup Penicillin amidase 3.5.1.11 Bacillus I Pharmaceutical Proteasei 3.4.21.14 Bacillus E +++ Detergent Pullulanasej 3.2.1.41 Klebsiella E Starch Fungal enzymes Amylase 3.2.1.1 Aspergillus E ++ Baking
Aminoacylase 3.5.1.14 Aspergillus I Pharmaceutical Glucoamylasek 3.2.1.3 Aspergillus E +++ Starch Catalase 1.11.1.6 Aspergillus I Food Cellulase 3.2.1.4 Trichoderma E Waste Dextranase 3.2.1.11 Penicillium E Food Glucose oxidase 1.1.3.4 Aspergillus I Food Lactasel 3.2.1.23 Aspergillus E Dairy Lipasee 3.1.1.3 Rhizopus E Food Rennetm 3.4.23.6 Mucor miehei E ++ Cheese Pectinasen 3.2.1.15 Aspergillus E ++ Drinks Pectin lyase 4.2.2.10 Aspergillus E Drinks Proteasem 3.4.23.6 Aspergillus E + Baking Raffinaseo 3.2.1.22 Mortierella I Food Yeast enzymes Invertasep 3.2.1.26 Saccharomyces I/E Confectionery Lactasel 3.2.1.23 Kluyveromyces I/E Dairy Lipasee 3.1.1.3 Candida E Food Raffinaseo 3.2.1.22 Saccharomyces I Food a The names in common usage are given. As most industrial enzymes consist of mixtures of enzymes, these names may vary from the recommended names of their principal component. Where appropriate, the recommended names of this principal component is given below. b The EC number of the principal component. c I - intracellular enzyme; E - extracellular enzyme. d +++ > 100 ton year-1; ++ > 10 ton year-1; + > 1 ton year-1; - < 1 ton year-1. e triacylglycerol lipase; f chymosin; g Endo-1,3(4)- -glucanase; h xylose isomerase; i subtilisin; j dextrin endo-1,6- -glucosidase; k glucan 1,4- -glucosidase; l -galactosidase; m microbial aspartic proteinase; n polygalacturonase; o -galactosidase; p -fructofuranosidase. In practice, the great majority of microbial enzymes come from a very limited number of genera, of which Aspergillus species, Bacillus species and Kluyveromyces (also called Saccharomyces) species predominate. Most of the strains used have either been employed by the food industry for many years or have been derived from such strains by mutation and selection. There are very few examples of the industrial use of enzymes having been developed for one task. Shining examples of such developments are the production of high fructose syrup using glucose isomerase and the use of pullulanase in starch hydrolysis.
Media for enzyme production Detailed description of the development and use of fermentors for the large-scale cultivation of microorganisms for enzyme production is outside the scope of this volume but mention of media use is appropriate because this has a bearing on the cost of the enzyme and because media components often find their way into commercial enzyme preparations. Details of components used in industrial scale fermentation broths for enzyme production are not readily obtained. This is not unexpected as manufacturers have no wish to reveal information that may be of technical or commercial value to their competitors. Also some components of media may be changed from batch to batch as availability and cost of, for instance, carbohydrate feedstock change. Such changes reveal themselves in often quite profound differences in appearance from batch to batch of a single enzyme from a single producer. The effects of changing feedstock must be considered in relation to downstream processing. If such variability is likely to significantly reduce the efficiency of the standard methodology, it may be economical to use a more expensive defined medium of easily reproducible composition. Clearly defined media are usually out of the question for large scale use on cost grounds but may be perfectly acceptable when enzymes are to be produced for high value uses, such as analysis or medical therapy where very pure preparations are essential. Less-defined complex media are composed of ingredients selected on the basis of cost and availability as well as composition. Waste materials and byproducts from the food and agricultural industries are often major ingredients. Thus molasses, corn steep liquor, distillers soluble and wheat bran is important components of fermentation media providing carbohydrate, minerals, nitrogen and some vitamins. Extra carbohydrate is usually supplied as starch, sometimes refined but often simply as ground cereal grains. Soybean meal and ammonium salts are frequently used sources of additional nitrogen. Most of these materials will vary in quality and composition from batch to batch causing changes in enzyme productivity. Often the enzyme may be purified several hundred-fold but the yield of the enzyme may be very poor, frequently below 10% of the activity of the original material (Table 2.2). In contrast, industrial enzymes will be purified as little as possible, only other enzymes and material likely to interfere with the process which the enzyme is to catalyze, will be removed. Unnecessary purification will be avoided as each additional stage is costly in terms of equipment, manpower and loss of enzyme activity. As a result, some commercial enzyme preparations consist essentially of concentrated fermentation broth, plus additives to stabilize the enzyme's activity. The content of the required enzyme should be as high as possible (e.g. 10% w/w of the protein) in order to ease the downstream processing task. This may be achieved by developing the fermentation conditions or, often more dramatically, by genetic engineering. It may well be economically viable to spend some time cloning extra copies of the required gene together with a powerful promoter back into the producing organism in order to get 'over-producers' (see Chapter protein engineering). It is important that the maximum activity is retained during the preparation of enzymes. Enzyme inactivation can be caused by heat, proteolysis, sub-optimal pH, oxidation, denaturants, irreversible inhibitors and loss of cofactors or coenzymes. Of this heat inactivation, this together with associated pH effects is probably the most significant. It is likely to occur during enzyme extraction and purification if insufficient cooling is available, but the problem is less when preparing thermophilic enzymes. Proteolysis is most likely to occur in the early stages of extraction and purification when the proteases responsible for protein turnover in living cells are still present. It is also the major reason for enzyme inactivation by microbial contamination. In their native conformations, enzymes have highly structured domains which are resistant to attack by proteases because many of the peptide bonds are mechanically inaccessible and because many proteases are highly specific. The chances of a susceptible peptide bond in a structured
domain being available for protease attack are low. Single 'nicks' by proteases in these circumstances may have little immediate effect on protein conformation and, therefore, activity. The effect, however, may severely reduce the conformational stability of the enzyme to heat or pH variation so greatly reducing its operational stability. If the domain is unfolded under these changed conditions, the whole polypeptide chain may be available for proteolysis and the same, specific, protease may destroy it. Clearly the best way of preventing proteolysis is to rapidly remove, or inhibit, protease activity. Before this can be achieved it is important to keep enzyme preparations cold to maintain their native conformation and slow any protease action that may occur. Some intracellular enzymes are used commercially without isolation and purification but the majority of commercial enzymes is either produced extracellularly by the microbe or plant or must be released from the cells into solution and further processed (Figure 2.1). Solid/liquid separation is generally required for the initial separation of cell mass, the removal of cell debris after cell breakage and the collection of precipitates. This can be achieved by filtration, centrifugation or aqueous biphasic partition. In general, filtration or aqueous biphasic systems are used to remove unwanted cells or cell debris whereas centrifugation is the preferred method for the collection of required solid material. Figure 2.1. Flow diagram for the preparation of enzymes. The main objective of the purification is To obtained maximum possible yield. It should have maximum possible purity. It should have maximum possible activity. Cell can be break down byfollowing methods 1. Osmotic shock 2. Enzymic lysis. 3. Homogenization 4. Ultracentrifugation. A. Cell breakage by osmotic shock Various intracellular enzymes are used in significant quantities and must be released from cells and purified (Table 2.1). The amount of energy that must be put into the breakage of cells depends much on the type of organism and to some extent on the physiology of the organism. Some types of cell are broken readily by gentle treatment such as osmotic shock (e.g. animal cells and some gram-negative bacteria such as Azotobacter species), whilst others are highly resistant to breakage. These include yeasts, green algae, fungal mycelia and some gram-positive bacteria which have cell wall and membrane structures capable of resisting internal osmotic pressures of around 20 atmospheres (2 MPa) and therefore have the strength, weight for weight, of reinforced concrete. Consequently a variety of cell disruption techniques have been developed involving solid or liquid shear or cell lysis. The rate of protein released by mechanical cell disruption is usually found to be proportional to the amount of releasable protein. (2.5) where P represents the protein content remaining associated with the cells, t is the time and k is a release constant dependent on the system. Integrating from P = Pm (maximum possible protein releasable) at time zero to P = Pt at time t gives (2.6) (2.7) As the protein released from the cells (Pr) is given by (2.8) the following equation for cell breakage is obtained (2.9) It is most important in choosing cell disruption strategies to avoid damaging the enzymes. The particular hazards to enzyme activity relevant to cell breakage are summarized in Table 2.3. The most significant of these, in general, are heating
and shear. ________________________________________ Table 2.3. Hazards likely to damage enzymes during cell disruption. Heat All mechanical methods require a large input of energy, generating heat. Cooling is essential for most enzymes. The presence of substrates, substrate analogues or polyols may also help stabilise the enzyme. Shear Shear forces are needed to disrupt cells and may damage enzymes, particularly in the presence of heavy metal ions and/or an air interface. Proteases Disruption of cells will release degradative enzymes which may cause serious loss of enzyme activity. Such action may be minimised by increased speed of processing with as much cooling as possible. This may be improved by the presence of an excess of alternative substrates (e.g. inexpensive protein) or inhibitors in the extraction medium. pH Buffered solutions may be necessary. The presence of substrates, substrate analogues or polyols may also help stabilise the enzyme. Chemical Some enzymes may suffer conformational changes in the presence of detergent and/or solvents. Polyphenolics derived from plants are potent inhibitors of enzymes. This problem may be overcome by the use of adsorbents, such as polyvinylpyrrolidone, and by the use of ascorbic acid to reduce polyphenol oxidase action. Oxidation Reducing agents (e.g. ascorbic acid, mercaptoethanol and dithiothreitol) may be necessary. Foaming The gas-liquid phase interfaces present in foams may disrupt enzyme conformation. Heavy-metal toxicity Heavy metal ions (e.g. iron, copper and nickel) may be introduced by leaching from the homogenisation apparatus. Enzymes may be protected from irreversible inactivation by the use of chelating reagents, such as EDTA. ________________________________________ Media for enzyme extraction will be selected on the basis of cost-effectiveness so will include as few components as possible. Media will usually be buffered at a pH value which has been determined to give the maximum stability of the enzyme to be extracted. Other components will combat other hazards to the enzyme, primarily factors causing denaturation (Table 2.3). B. Use of enzymic lytic methods The breakage of cells using non-mechanical methods is attractive in that it offers the prospects of releasing enzymes under conditions that are gentle, do not subject the enzyme to heat or shear, may be very cheap, and are quiet to the user. The methods that are available include osmotic shock, freezing followed by thawing, cold shock, desiccation, enzymic lysis and chemical lysis. Each method has its drawbacks but may be particularly useful under certain specific circumstances. Certain types of cell can be caused to lyse by osmotic shock. This would be a cheap, gentle and convenient method of releasing enzymes but has not apparently been used on a large scale. Some types of cell may be caused to autolyse, in particular yeasts and Bacillus species. Yeast invertase preparations employed in the industrial manufacture of invert sugars are produced in this manner. Autolysis is a slow process compared with mechanical methods, and microbial contamination is a potential hazard, but it can be used on a very large scale if necessary. Where applicable, desiccation may be very useful in the preparation of enzymes on a large scale. The rate of drying is very important in these cases, slow methods being preferred to rapid ones like lyophilisation. Enzymic lysis using added enzymes has been used widely on the laboratory scale but is less popular for industrial purposes. Lysozyme, from hen egg-white, is the only lytic enzyme available on a commercial scale. It has often used to lyse Gram positive bacteria in an hour at about 50,000 U Kg-1 (dry weight). The chief objection to its use on a large scale is its cost. Where costs are reduced by the use of the relatively inexpensive, Lysozyme-rich, dried egg white, a major separation problem may be introduced. Yeast-lytic enzymes from Cytophaga species
have been studied in some detail and other lytic enzymes are under development. If significant markets for lytic enzymes are identified, the scale of their production will increase and their cost is likely to decrease.Lysis by acid, alkali, surfactants and solvents can be effective in releasing enzymes, provided that the enzymes are sufficiently robust. Detergents, such as Triton X-100, used alone or in combination with certain chaotropic agents, such as guanidine HCl, are effective in releasing membrane-bound enzymes. However, such materials are costly and may be difficult to remove from the final product. c. Ultrasonic cell disruption The treatment of microbial cells in suspension with inaudible ultrasound (greater than about 12 kHz) results in their inactivation and disruption. Ultrasonication utilises the rapid sinusoidal movement of a probe within the liquid. It is characterised by high frequency (12 kHz - 1 MHz), small displacements (less than about 50 m), moderate velocities (a few m s-1), steep transverse velocity gradients (up to 4,000 s-1) and very high acceleration (up to about 20,000 g). Ultrasonication produces cavitation phenomena when acoustic power inputs are sufficiently high to allow the multiple production of microbubbles at nucleation sites in the fluid. The bubbles grow during the rarefying phase of the sound wave, then are collapsed during the compression phase. On collapse, a violent shock wave passes through the medium. The whole process of gas bubble nucleation, growth and collapse due to the action of intense sound waves is called cavitation. The collapse of the bubbles converts sonic energy into mechanical energy in the form of shock waves equivalent to several thousand atmospheres (300 MPa) pressure. This energy imparts motions to parts of cells which disintegrate when their kinetic energy content exceeds the wall strength. An additional factor which increases cell breakage is the microstreaming (very high velocity gradients causing shear stress) which occur near radially vibrating bubbles of gas caused by the ultrasound. Much of the energy absorbed by cell suspensions is converted to heat so effective cooling is essential. The amount of protein released by sonication has been shown to follow Equation 2.9. The constant (k) is independent of cell concentrations up to high levels and approximately proportional to the input acoustic power above the threshold power necessary for cavitation. Disintegration is independent of the sonication frequency except insofar as the cavitation threshold frequency depends on the frequency. Equipment for the large-scale continuous use of ultrasonic has been available for many years and is widely used by the chemical industry but has not yet found extensive use in enzyme production. Reasons for this may be the conformational lability of some (perhaps most) enzymes to sonication and the damage that they may realize though oxidation by the free radicals, singlet oxygen and hydrogen peroxide that may be concomitantly produced. Use of radical scavengers (e.g. N2O) have been shown to reduce this inactivation. As with most cell breakage methods, very fine cell debris particles may be produced which can hinder further processing. Sonication remains, however, a popular, useful and simple small-scale method for cell disruption. d. Cell lysis by High pressure homogenisers Various types of high pressure homogeniser are available for use in the food and chemicals industries but the design which has been very extensively used for cell disruption is the Manton-Gaulin APV type homogeniser. This consists of a positive displacement pump which draws cell suspension (about 12% w/v) through a check valve into the pump cylinder and forces it, at high pressures of up to 150 MPa (10 tons per square inch) and flow rates of up to 10,000 L hr-1, through an adjustable discharge valve which has a restricted orifice (Figure 2.5). Cells are subjected to impact, shear and a severe pressure drop across the valve but the precise mechanism of cell disruption is not clear. The main disruptive factor is the pressure applied and consequent pressure drop across the valve. This causes the impact and shear stress which are proportional to the operating pressure.
Figure 2.2. A cross-section through the Manton-Gaulin homogeniser valve, showing the flow of material. The cell suspension is pumped at high pressure through the valve impinging on it and the impact ring. The shape of the exit nozzle from the valve seat varies between models and appears to be a critical determinant of the homogenisation efficiency. The model depicted is the 'CD Valve' from APV Gaulin. ________________________________________ As narrow orifices which are vulnerable to blockage are key parts of this type of homogeniser, it is unsuitable for the disruption of mycelial organisms but has been used extensively for the disruption of unicellular organisms. The release of proteins can be described by Equation 2.9 but normally a similar relationship is used where the time variable is replaced by the number of passes (N) through the homogeniser. (2.10) In the commonly-used operating range with pressures below about 75 MPa, the release constant (k) has been found to be proportional to the pressure raised to an exponent dependent on the organism and its growth history (e.g. k=k'P2.9 in Saccharomyces cerevesiae and k=k'P2.2 in Escherichia coli, where P represents the operating pressure and k' is a rate constant). Different growth media may be selected to give rise to cells of different cell wall strength. Clearly, the higher the operating pressure, the more efficient is the disruption process. The protein release rate constant (k) is temperature dependent, disruption being more rapid at higher temperatures. In practice, this advantage cannot be used since the temperature rise due to adiabatic compression is very significant so samples must be pre-cooled and cooled again between multiple passes. At an operating pressure of 50 MPa, the temperature rise each pass is about 12 deg. C. In addition to the fragility of the cells, the location of an enzyme within the cells can influence the conditions of use of an homogeniser. Unbound intracellular enzymes may be released by a single pass whereas membrane bound enzymes require several passes for reasonable yields to be obtained. Multiple passes are undesirable because, of course, they decrease the throughput productivity rate and because the further passage of already broken cells results in fine debris which is excessively difficult to remove further downstream. Consequently, homogenisers will be used at the highest pressures compatible with the reliability and safety of the equipment and the temperature stability of the enzyme(s) released. High pressure homogenisers are acceptably good for the disruption of unicellular organisms provided the enzymes needed are not heat labile. The shear forces produced are not capable of damaging enzymes free in solution. The valve unit is prone to erosion and must be precision made and well maintained. e. Use of bead mills for cell lysis When cell suspensions are agitated in the presence of small steel or glass beads (usually 0.2 -.1.0 mm diameter) they are broken by the high liquid shear gradients and collision with the beads. The rate and effectiveness of enzyme release can be modified by changing the rates of agitation and the size of the beads, as well as the dimensions of the equipment. Any type of biomass, filamentous or unicellular, may be disrupted by bead milling but, in general, the larger sized cells will be broken more readily than small bacteria. For the same volume of beads, a large number of small beads will be more effective than a relatively small number of larger beads because of the increased likelihood of collisions between beads and cells. Bead mills are available in various sizes and configurations from the Mickle shaker which has a maximum volume of about 40 ml to continuous process equipment capable of handling up to 200 Kg wet yeast or 20 Kg wet bacteria each hour. The bead mills that have been studied in most detail are the Dyno-Mill and the NetschMolinex agitator, both of which consist of a cylindrical vessel containing a motor-driven central shaft equipped with impellers of different types. Both can be operated continuously, being equipped with devices which retain the beads within the milling chamber. Glass Ballotini or stainless steel balls are used, the size
range being selected for most effective release of the enzyme required. Thus 1 mm diameter beads are satisfactory for the rapid release of periplasmic enzymes from yeast but 0.25 mm diameter beads must be used, for a longer period, to release membrane-bound enzymes from bacteria. The kinetics of protein release from bead mills follows the relationship given by Equation 2.9 with respect to the time (t) that a particle spends in the mill. Unfortunately, however well designed these mills are, when continuously operated there will be a significant amount of backmixing which reduces the efficiency of the protein released with respect to the average residence time () . This is more noticeable at low flow rates (high average residence times) and when the proportion of protein released is high. It may be counteracted by designing the bead mill to encourage plug flow characteristics. Under these circumstances the relationship can be shown to be (2.11) where i represents the degree of backmixing (i.e. i = 0 under ideal plug flow conditions and i = 1 for ideal complete backmixing). Equation 2.11 reduces to give the simplified relationship of Equation 2.9 at low (near zero) values of i. In addition to bead size, the protein release rate constant (k) is a function of temperature, bead loading, impeller rotational speed and cell loading. Impeller speeds can be increased with advantage until bead breakage becomes significant but heat generation will also increase. At a constant impeller speed, the efficiency of the equipment declines with throughput as the degree of backmixing increases. There will be an optimum impeller tip speed at which the increases in disruption are balanced by increases in backmixing. In general, increased bead loading increases the rate of protein release but also increases the production of heat and the power consumption. Heat production is the major problem in the use of bead mills for enzyme release, particularly on a large (e.g. 20 liters) scale. Smaller vessels may be cooled adequately through cooling jackets around the bead chamber but larger mills require cooling through the agitator shaft and impellers. However, if cooling is effective there is little damage to the enzymes released. 2.6 Method of separation of enzyme from lysed cell; Centrifugation B. ULTRACENTRIFUGATION This technique is used to investigate the different parameter of the molecule such as molecular mass, shape, and density. In this process gravity plays most important role. Therefore, the basis of centrifugation separation techniques therefore is to exert the lager force than does the gravitational earth, thus increasing the rate at which the particle settles. The technique can be divided into two types: 1 preparative centrifugation: for separation of the whole cells, subcellular organelles, plasma membranes, polysomes, ribosome, chromatin, nucleic acids, lipoprotein and viruses 2. Analytical centrifugation: it is devoted to study of pure or virtually pure or macromolecular or particles. They are related with the study of macromolecular structure rather than the collection of particle fractions. Principles: Rate depends on the centrifugal field G directed radially outward (angular velocity) G = r 2 =2/60 revolution min-1 ultracentrifuge have maximum speed of 600,000 g. the chamber is surrounded by the refrigerated, sealed and evacuated to minimized excess rise in temperature. Note that there must be equal loading of sample. For safety purpose ultracentrifuge are always enclosed in heavy armour plating. This is type of preparative
ultracentrifuge, better for the deproteinisation of physiological fluids for amino acid analysis. Another type of ultracentrifuge is analytical type. It has speed 70,000 rev per min (500,000 g). its fabrication is of same type. It can also measure the refractive index, and is equipped with the ultraviolet light absorption system. Light of suitable wavelength is passed through moving analytical cell containing the solution under analysis e.g. protein or nucleic acid and intensity of light is measured on the photographic plate. Optical system records the change in refractive index. The Schleiren optical system plots the refractive index gradient against distance along the analytical cell, which makes it useful in analysis in the locating in boundaries in sedimentation velocity measurements. A. Differential centrifugation This method is based upon the difference in the sedimentation rate of particles of different size and density. In centrifugation the larger particle are sedimented first. The particle having the same mass but different density, the denser particle is sedimented first and less dense will sediment later. Particle having similar density can be separated by the differential centrifugation or the rate zonal method. In differentiated centrifugation, the particle to be separated is divided centrifugally into a number of fractions by increasing the applied centrifugal field. After the centrifugation, pellet and supernatant are separated, pellet is washed several time and again centrifugation is done. The particle moves against respective sedimentation rates. Centrifugation is continued long enough to pellet all the largest class of particles, the resulting supernatant then being centrifuged at higher speed to separate medium sized particle and so on. However, since particles of varying sizes and densities were distributed homogeneously at the start of centrifugation, so pellet will not be homogeneous but will contain a mixture of all the sediment components. Particle separation by the rate zonal technique is based on differences in the size, shape, and density of the particle. Most of the biological particles having same size are having narrower density range. So , separation of similar particle by the rate zonal technique is based mainly upon differences in their sizes and can not be separated easily like mitochondria, lysosome, peroxisomes. The technique has been used for the separation of enzymes hormones and RNA and DNA hybrids, ribosomal subunits, sub cellular organelles. There are two type of density gradient centrifugation, --1. The rate zonal technique and the isopycnic (equal density) Isopycnic centrifugation depends solely upon the buoyant density of the particles and not its shape or size and is independent of time the size of the particle affecting only the rate at which it reaches its isopycnic position in the gradient. The technique is useful in separating the particle of same size but differing in density. 1Svedberg = 10-13 second Centrifugation separates on the basis of the particle size and density difference between the liquid and solid phases. Sedimentation of material in a centrifugal field may be described by (2.11) where v is the rate of sedimentation, d is the particle diameter, rs is the particle density, rl is the solution density is the angular velocity in radians s1, r is the radius of rotation, is the kinematic viscosity, Fs is a correction factor for particle interaction during hindered settling and is a shape factor (=1 for spherical particles). Fs depends on the volume fraction of the solids present; approximately equaling 1, 0.5, 0.1 and 0.05 for 1%, 3%, 12% and 20% solids volume fraction respectively. Only material which reaches a surface during the flow through continuous centrifuges will be removed from the centrifuge
feedstock, the efficiency depending on the residence time within the centrifuge and the distance necessary for sedimentation (D). This residence time will equal the volumetric throughput () divide by the volume of the centrifuge (V). The maximum throughput of a centrifuge for efficient use is given by (2.12) The efficiency of the process is seen to depend on the solids volume fraction, the effective clarifying surface (V/D) and the acceleration factor (2r/g, where g is the gravitational constant, 921 cm s-2; a rotor of radius 25 cm spinning at 1 rev s-1 has an acceleration factor of approximately 1 G). Low acceleration factors of about 1 500 g may be used for harvesting cells whereas much higher acceleration factors are needed to collect enzyme efficiently. The product of these factors (2rV/gD) is called the sigma factor and is used to compare centrifuges and to assist scale-up. Laboratory centrifuges using tubes in swing-out or angle head rotors have high angular velocity and radius of rotation (r) but small capacity (V) and substantial sedimentation distance (D). This type of design cannot be scaled-up safely, primarily because the mechanical stress on the centrifuge head increases with the square of the radius, which must increase with increasing capacity. For large-scale use, continuous centrifuges of various types are employed (Figure 2.2). These allow the continuous addition of feedstock, the continuous removal of supernatant and the discontinuous, semicontinuous or continuous removal of solids. Where discontinuous or semicontinuous removal of precipitate occurs, the precipitate is flushed out by automatic discharge systems which cause its dilution with water or medium and may be a problem if the precipitate is required for further treatment. Centrifugation is the generally preferred method for the collection of enzyme-containing solids as it does not present a great hazard to most enzymes so long as foam production, with consequent enzymic inactivation, is minimised. centrifuges are long and thin enabling rapid acceleration and deceleration, minimizing the down-time required for the removal of the sedimented solids. Here the radius and effective liquid thickness are both small allowing a high angular velocity and hence high centrifugal force; small models can be used at acceleration factors up to 50,000 g, accumulating 0.1 Kg of wet deposit whereas large models, designed to accumulate up to 5 Kg of deposit, are restricted to 16,000 g. The capacities of these centrifuges are only moderate.Multichamber discstack centrifuges, originally designed (by Westfalia and Alpha-Laval) for cream separation, contain multiple coned discs in a stack which are spun and on which the precipitate collects. They may be operated either semi-continuously or, by using a centripetal pressurising pump within the centrifuge bowl which forces the sludge out through a valve, continuously. The capacity and radius of such devices are large and the thickness of liquid is very small, due to the large effective surface area. The angular velocity, however, is restricted giving a maximum acceleration factor of about 2,000 g. A different design which is rather similar in principle is the solid bowl scroll centrifuge in which an Archimedes' screw collects the precipitate so that fluid and solids leave at opposite ends of the apparatus. These can only be used at low acceleration (about 3,000 g) so they are suitable only for the collection of comparatively large particles. Although many types of centrifuge are available, the efficient precipitation of small particles of cell debris can be difficult, sometimes near-impossible. Clearly from Equation 2.2, the efficiency of centrifugation can be improved if the particle diameter (d) is increased. This can be done either by coagulating or flocculating particles. Coagulation is caused by the removal of electrostatic charges (e.g. by pH change) and allowing particles to adhere to each other. Flocculation is achieved by adding small amounts of high-molecular-weight charged materials which bridge oppositely-charged particles to produce a loose aggregate which may be readily removed by centrifugation or filtration. Flocculation and coagulation are cheap and effective aids to precipitating or otherwise harvesting whole cells, cell debris or soluble proteins but, of course, it is essential that
the agents used must not inhibit the target enzymes. It is important to note that the choice of flocculants is determined by the pH and ionic strength of the solution and the nature of the particles. Most flocculants have very definite optimum concentrations above which further addition may be counter-effective. Some flocculants can be rapidly ruined by shear. A comparatively recent introduction designed for the removal of cell debris is a moderately hydrophobic product in which cellulose is lightly derivatised with diethylaminoethyl functional groups. This material (Whatman CDR; cell debris remover) is inexpensive (essential as it is not reusable), binds to unwanted negatively charged cell constituents, acts as a filter aid and may be incinerated to dispose of hazardous 2.7 Filtration of enzyme after separation Filtration separates simply on the basis of particle size. Its efficiency is limited by the shape and compressibility of the particles, the viscosity of the liquid phase and the maximum allowable pressures. Large-scale simple filtration employs filter cloths and filter aids in a plate and frame press configuration, in rotary vacuum filters or centrifugal filters (Figure 2.3). The volumetric throughput of a filter is proportional to the pressure (P) and filter area (AF) and inversely proportional to the filter cake thickness (DF) and the dynamic viscosity (2.13) Where, k is a proportionality constant dependent on the size and nature of the particles. For very small particles k depends on the fourth power of their diameter. Filtration of particles that are easily compressed leads to filter blockage and the failure of Equation 2.3 to describe the system. Under these circumstances a filter aid, such as celite, is mixed with the feedstock to improve the mechanical stability of the filter cake. Filter aids are generally used only where the liquid phase is required as they cause substantial problems in the recovery of solids. They also may cause loss of enzyme activity from the solution due to physical hold-up in the filter cake. It is often difficult for a process development manager to decide whether to attempt to recover enzyme trapped in this way. Problems associated with the build-up of the filter cake may also be avoided by high tangential flow of the feedstock across the surface of the filter, a process known as crossflow microfiltration (Figure 2.4). This method dispenses with filter aids and uses special symmetric microporous membrane assemblies capable of retaining particles down to 0.1 - 1 m diameter (cf. Bacillus diameter of about 2 m). ________________________________________ Figure 2.3. The basic design of the rotary vacuum filter. The suspension is sucked through a filter cloth on a rotating drum. This produces a filter cake which is removed with a blade. The filter cake may be rinsed during its rotation. These filters are generally rather messy and difficult to contain making them generally unsuitable for use in the production of toxic or recombinant DNA products. There have been recent developments that improve their suitability, however, such as the Disposable Rotary Drum Filter. ________________________________________ A simple and familiar filtration apparatus is the perforate bowl centrifuge or basket centrifuge, in effect a spin drier. Cell debris is collected on a cloth with, or without, filter aid and can be skimmed off when necessary using a suitable blade. Such centrifugal filters have a large radius and effective liquid depth, allowing high volumes. However, safety decrees that the angular velocity must be low and so only large particles (e.g. plant material) can be removed satisfactorily. ________________________________________ Figure 2.4. Principles of (a) dead-end filtration and (b) cross-flow filtration.
In dead-end filtration the flow causes the build-up of the filter cake, which may prevent efficient operation. This is avoided in cross-flow filtration where the flow sweeps the membrane surface clean. ________________________________________ Chapter-10 Separation in Aqueous biphasic systems The 'incompatibility' of certain polymers in aqueous solution was first noted by Beijerinck in 1296. In this case two phases were formed when agar was mixed with soluble starch or gelatin. Since then, many two phase aqueous systems have been found; the most thoroughly investigated being the aqueous dextran-polyethylene glycol system (e.g. 10% polyethylene glycol 4000/2% dextran T500), where dextran forms the more hydrophilic, denser, lower phase and polyethylene glycol the more hydrophobic, less dense, upper phase. Aqueous three phase systems are also known. Phases form when limiting concentrations of the polymers are exceeded. Both phases contain mainly water and are enriched in one of the polymers. The limiting concentrations depend on the type and molecular weight of the polymers and on the pH, ionic strength and temperature of the solution. Some polymers form the upper hydrophobic phase in the presence of fairly concentrated solutions of phosphates or sulphate (e.g. 10% polyethylene glycol 4000/12.5% potassium phosphate buffer). A drawback to the useful dextran/polyethylene glycol system is the high cost of the purified Dextran used. This has been alleviated by the use of crude unfractionated dextran preparations, much cheaper hydroxypropyl starch derivatives and salt-containing biphasic systems. Aqueous biphasic systems are of considerable value to biotechnology. They provide the opportunity for the rapid separation of biological materials with little probability of denaturation. The interfacial tension between the phases is very low (i.e. about 400-fold less than that between water and an immiscible organic solvent), allowing small droplet size, large interfacial areas, efficient mixing under very gentle stirring and rapid partition. The polymers have a stabilizing influence on most proteins. A great variety of separations have been achieved, by far the most important being the separation of enzymes from broken crude cell material. Separation may be achieved in a few minutes, minimizing the harmful action of endogenous proteases. The systems have also been used successfully for the separation of different types of cell membranes and organelles, the purification of enzymes and for extractive bioconversions. Continuous liquid twophase separation is easier than continuous solid/liquid separation using equipment familiar from immiscible solvent systems, for example disc-stack centrifuges and counter-current separators. Such systems are readily amenable to scale-up and may be employed in continuous enzyme extraction processes involving some recycling of the phases. Cells, cell debris proteins and other material distribute themselves between the two phases in a manner described by the partition coefficient (P) defined as. --------------------(2.14) Where Ct and Cb represent the concentrations in the top and bottom phases respectively. The yield and efficiency of the separation is determined by the relative amounts of material in the two phases and therefore depends on the volume ratio (Vt/Vb). The partition coefficient is exponentially related to the surface area (and hence molecular weight) and surface charge of the particles in addition to the difference in the electrical potential and hydrophobicity of the phases. It is not generally very sensitive to temperature changes. This means that proteins and larger particles are normally partitioned into one phase whereas smaller molecules are distributed more evenly between phases. A partition coefficient of greater than 3 is required if usable yields are to be achieved by a single extraction process. Typical partition coefficients for proteins are 0.01-100 whereas the partition coefficients for cells and cell debris are effectively zero. The influence of pH and salts on protein partition is complex, particularly when phosphate buffers are present. A given protein distributes differently between the phases at different pH's and ionic strength but the presence of phosphate ions
affect the partition coefficient in an anomalous fashion because these ions distribute themselves unequally resulting in electrostatic potential (and pH) differences. This means that systems may be 'tuned' to enrich an enzyme in one phase, ideally the upper phase with cell debris and unwanted enzymes in the lower phase. An enzyme may be extracted from the upper (polyethylene glycol) phase by the addition of salts or further polymer, generating a new biphasic system. This stage may be used to further purify the enzyme. A powerful modification of this technique is to combine phase partitioning and affinity partitioning. Affinity ligand (e.g. triazine dyes) may be coupled to either polymer in an aqueous biphasic system and thus greatly increase the specificity of the extraction. Phases form when limiting concentrations of the polymers are exceeded. Both phases contain mainly water (typically 70-90% w/w water) and are enriched in one of the polymers. The limiting concentrations depend on the type and molecular weight of the polymers and on the pH, ionic strength and temperature of the solution. Some polymers form a two-phase system by themselves; PEG forming the upper morehydrophobic phase in the presence of fairly concentrated solutions of citrates, phosphates or sulfates or at higher temperatures (see below). Such aqueous liquidliquid two-phase systems are finding increasing use in the extractive separation of labile biomolecules such as proteins, offering mild conditions due to the low interfacial tension between the phases (i.e. about 400-fold less than that between water and an immiscible organic solvent) allowing small droplet size, large interfacial areas, efficient mixing under very gentle stirring and rapid partition. The polymers also have a stabilizing influence on most proteins. A great variety of separations have been achieved, by far the most important being the separation of enzymes from broken crude cell material. Separation may be achieved in a few minutes, minimizing the harmful action of endogenous proteases. The systems have also been used successfully for the separation of different types of cell membranes, organelles and actinide ions, the purification of enzymes, extractive bioconversions. Although sometimes perceived as due to polymer incompatibilities, the properties of these biphasic systems can be mainly attributed to incompatibility between aqueous pools of low and higher density water. Each phase may be considered as a different, although aqueous, solvent with properties determined by its structuring. PEG usually has a far higher concentration in the upper (low-density) phase of such solutions in spite of its inherent density being greater than water. These, together with the properties of this PEG phase encourage the belief that it creates a predominantly low-density water environment due to its partially hydrophobic character, in turn mainly determined by the methylene groups. Further proof of this may be seen by use of microwave dielectric measurements, which show the water surrounding PEG to be ordered, whereas that surrounding more hydrophilic polymers is disordered [332]. Also, the dissolution of PEG is exothermic (and increasingly exothermic with PEG size), in line with a shift in the ES CS equilibrium towards the more ordered ES structure. It is interesting and perhaps not simply fortuitous that the diameter (4.9 ) of the favored PEG helix (formed by trans, gauche, trans links across the C-O-C-C, O-C-C-O, C-C-O-C bonds) is the same as the diameter of the spines of the ES water cluster (4.7 ) formed by pentagonal boxes, the ether (O-C-C-O) distances (2.22 ) are close to the OO distances (2.24 ) in water and the next ether (O-C-C-O-C-C-O) (5.6 ) distances are close to the next vertex distance on opposite sides of the pentagonal boxes (5.4 ).a Model building shows that optimum hydrogen bonding would tend to distort this PEG helix, however. The strongly-held hydration, as determined by viscosity, increases from two molecules of water per PEG monomer at very low polymerization (tetramer) to 5 molecules of water per PEG monomer for 45-mer , showing that the extent of water clustering increases with PEG size. The partitioning of proteins into the hydrophobic PEG phase shows great sensitivity to the protein's surface hydrophobicity (partition increasing with surface hydrophobicity) and also depends on the PEG size; increasing with PEG molecular mass , in line with the extent of
water clustering. Increasing PEG size and concentration both increase the proteins' effective hydration as the PEG is excluded from the proteins' surface. However, when the PEG phase becomes too ordered (e.g. at higher PEG size) partitioned proteins are excluded due to the reduced available water content. An interesting and revealing phenomenon occurs in PEG solutions as the temperature is raised; the solution at low temperatures separates into two phases (PEG-rich and PEG-poor) at higher temperature (separating at the cloud-point) and reverts to a single phase at even higher temperatures. This may be explained as the PEG creating a low-density water environment with decreased entropy. At low temperatures a solution is formed due to the enthalpy of hydrogen bonding between the PEG and the water more than compensating for the entropy lost in forming the low-density water. This entropy loss is required, due to the hydrophobicity of the methylene groups, but is not great as the water is somewhat ordered already at lower temperatures. At the cloud point, the entropy cost is greater as the water is no longer naturally as structured, and two phases develop. The stronger hydrogen bonding in D2O, relative to H2O, is expected to raise this cloudpoint. At higher temperatures still, the water possesses excess energy and cannot be structured by the PEG. This reduces the entropic cost, so allowing a solution to form once more. Anions have a distinct effect on the cloud point in line with the Hofmeister Series (cloud point lowering: SCN- < I- < Br- < Cl- < F- < OH- < SO42- < HPO42- < CO32- < PO43-); the greater lowering of the cloud point is in line with greater surface charge density , stronger hydration, greater tendency to avoid low-density water and the greater destruction of the natural structuring of the water. A oppositely-ordered compensating effect on the cloud point has been recognized due to binding of the anions to the polymer surface. This tends to raise the cloudpoint at lower salt concentrations as the bound salt increases the polymer net charge and, hence, solubility. The relative effect of the ions is the reverse of the Hofmeister series just given with weakly hydrated ions binding best, i.e. SCN- having the greatest effect and ionic kosmotropes below Cl- having negligible effect. Cations have a lesser but opposite effect to anions with chaotropes (e.g. NH4+) tending to lower the cloud point but kosmotropes (e.g. Li+) raising it. Exceptionally, however, some di- and trivalent cations such as Mg2+ and Zn2+ act counter to their normal Hofmeister behavior, due presumably to their specific chelation to oxygen atoms in the PEG molecules. Anions and cations distribute themselves differently between the phases depending on their affinity for low or higher density water but with the requirements that the phases be electrically neutral and iso-osmotic, so producing an interfacial potential difference, which may aid the partitioning of charged biomolecules. Thus sulfate and phosphate ions prefer the bottom phase and, as a consequence, negatively charged proteins are partitioned into the upper PEG phase, so allowing more sulfate or phosphate ions to partition into their preferred lower phase. Preference for the PEG-rich or PEG-poor phase is related to the Hofmeister Series for the structuring ability of the salts, particularly the anions (e.g. preference for PEG-rich phase: I- > Br- > Cl- > F- > SO42-; Cs+ > Na+ > Ba2+ > Ca2+; preference for PEG-poor phase: SO42- > F- > Cl- > Br- > I-). A similar Hofmeister Series effect is noticed intensifying the incompatibility between two polymers such as polyethylenimine-PEG, or dextran-PEG, by increasing the concentration of strongly hydrated (CS-forming) anions, such as sulfate. a Note that the polymers formed with either one (-O-C-O-C-O-) or three (-O-C-C-CO-C-C-C-O-) methylene groups between the oxygen atoms are both insoluble in water. The reason however is not so much that the OO distances (2.12 and 4.79 respectively) fit less well with the water cluster spacing but rather that the molecules form almost-linear extended (rather than helical) chains with a pronounced hydrophobic character that have strong intra-molecular attraction. 2.9 Preparation of enzymes from clarified solution; Ultrafiltration In many cases, especially when extracellular enzymes are being prepared for sale,
the clarified solution is simply concentrated, preservative materials added, and sold as a solution or as a dried preparation. The concentration process chosen will be the cheapest which is compatible with the retention of enzyme activity. For some enzymes rotary evaporation can be considered, followed if necessary by spray drying. The most popular method, though, is ultrafiltration, whereby water and low molecular weight materials are removed by passage through a membrane under pressure, enzyme being retained. Ultrafiltration differs from conventional filtration and microfiltration with respect to the size of particles being retained (< 50 nm diameter). It uses asymmetric microporous membranes with a relatively dense but thin skin, containing pores, supported by a coarse strong substructure. Membranes possessing molecular weight cut-offs from 1000 to 100,000 and usable at pressure up to 2 MPa are available. There are number types of apparatus available. Stirred cells represent the simplest configuration of ultrafiltration cell. The membrane rests on a rigid support at the base of a cylindrical vessel which is equipped with a magnetic stirrer to combat concentration polarization. It is not suitable for large scale use but is useful for preliminary studies and for the concentration of laboratory column eluates. Various large-scale units are available in which membranes are formed into wide diameter tubes (1 - 2 cm diameter) and the tubes grouped into cartridges. These are not as compact as capillary systems (area/volume about 25 m1) and are very expensive but are less liable to blockage by stray large particles in the feedstream. Cheaper thin-channel systems are available (area/volume about 500 m-1) which use flat membrane sandwiches in filter press arrangements of various designs chosen to produce laminar flow across the membrane and minimise concentration polarization. Capillary membranes represent a relatively cheap and increasingly popular type of ultrafiltration system which uses micro-tubular membranes 0.2 - 1.1 mm diameter and provides large membrane areas within a small unit volume (area/volume about 1000 m-1). Membranes are usually mounted into modules for convenient manipulation. This configuration of membranes can be scaled up with ease. Commercial models are available that give ultrafiltration rates of up to 600 L hr-1. The steady improvement in the performance, durability and reliability of membranes has been a boon to enzyme technologists, encouraging wide use of the various ultrafiltration configurations. Problems with membrane blockage and fouling can usually be overcome by treatment of membranes with detergents, proteases or, with care, acids or alkalis. The initial cost of membranes remains considerable but modern membranes are durable and cost-effective. Ultrafiltration, done efficiently, results in little loss of enzyme activity. However, some configurations of apparatus, particularly in which solutions are recycled, can produce sufficient shear to damage some enzymes.
Chapter-10 Concentration and purification of enzyme Concentration by precipitation Precipitation of enzymes is a useful method of concentration and is ideal as an initial step in their purification. It can be used on a large scale and is less affected by the presence of interfering materials than any of the chromatographic methods described later. There are method of salting in and salting out. Salting in is the method in which ammonium sulphate is used for dissolving the protein. Note that protein dissolves least at its isoelectric point. On increasing the ammonium sulphate concentration further proteins starts precipitating this phenomenon is called as salting out. All this process must be done in the ice cold solution so that no protein can denature. Salting out of proteins is done by use of ammonium sulphate, is one of the best known and used methods of purifying and concentrating enzymes, particularly at the laboratory scale. Increases in the ionic strength of the solution cause a reduction in the repulsive effect of like charges between identical molecules of a protein. It also reduces the forces holding the salvations shell around the protein molecules. When these forces are sufficiently reduced, the protein will precipitate; hydrophobic proteins precipitating at lower salt concentrations than hydrophilic proteins. Ammonium sulphate is convenient and effective because of its high solubility, cheapness, lack of toxicity to most enzymes and its stabilizing effect on some enzymes (see Table 2.4). Its large-scale use, however, is limited as it is corrosive except with stainless steel, it forms dense solutions presenting problems to the collection of the precipitate by centrifugation, and it may release gaseous ammonia, particularly at alkaline pH. The practice of using ammonium sulphate precipitation is more straightforward than the theory. Reproducible results can only be obtained provided the protein concentration, temperature and pH are kept constant. The concentration of the salt needed to precipitate an enzyme will vary with the concentration of the enzyme. However, fractionation of protein mixtures by the stepwise increase in the ionic strength can be a very effective way of partly purifying enzymes. The solubility of an enzyme can be described by the equation (2.11) where S is the enzyme solubility, Kintercept is the intercept constant, Ksalt is the salting out constant and T is the ionic strength which is proportional to the concentration of a precipitating salt. Kintercept is independent of the salt used but depends on the pH, temperature, enzyme and the other components in the solution. Ksalt depends on both the enzyme required and the salt used but is largely independent of other factors. This equation (2.11) may also be used to give the minimum salt concentration necessary before enzyme will start to
precipitate; the concentration change necessary to precipitate the enzyme varying according to the magnitude of the salting out constant. Some enzymes do not survive ammonium sulphate precipitation. Other salts may be substituted but the more favored alternative is to use organic solvents such as methanol, ethanol, propan-2-ol and acetone. These act by reducing the dielectric of the medium and consequently reducing the solubility of proteins by favoring protein-protein rather than protein-solvent interactions. Organic solvents are not widely used on a large scale because of their cost, their flammability, and the tendency of proteins to undergo rapid denaturation by these solvents if the temperature is allowed to raise much above 0C. On safety grounds when organic solvents are used, special flameproof laboratory areas are used and temperatures maintained below their flashpoints. Except when enzymes are presented for sale as ammonium sulphate precipitates, the precipitating salt or solvent must be removed. This may be done by dialysis, Ultrafiltration or by using a desalting column of, for instance, Sephadex G-25. A. Nucleic acid removal Intracellular enzyme preparations contain nucleic acids which can give rise to increased viscosity interfering with enzyme purification procedures, in particular ultrafiltration. Some organisms contain sufficient nuclease activity to eliminate this problem but, otherwise, the nucleic acids must be removed by precipitation or degraded by the addition of exogenous nucleases. Ammonium sulphate precipitation can be effective in removing nucleic acids but will remove some protein at the same time. Various more specific precipitants have been used, usually positivelycharged materials which form complexes with the negatively-charged phosphate residues of the nucleic acids. These include, in order of roughly decreasing effectiveness, polyethyleneimine, the cationic detergent cetyltrimethyl ammonium bromide, streptomycin sulphate and protamine sulphate. All of these are expensive and possibly toxic, particularly streptomycin sulphate. Also, they may complex undesirably with certain enzymes. They may be necessary, however, where possible contamination of the enzyme product must be avoided, such as in the preparation of restriction endonucleases. Otherwise, treatment with bovine pancreatic nucleases is probably the most cost-effective method of nucleic acid removal. B. Heat treatment In many cases, unwanted enzyme activities may be removed by heat treatment. Different enzymes have differing susceptibility to heat denaturation and precipitation. Where the enzyme required is relatively heat-stable this allows its easy and rapid purification in terms of enzymic activity. For such enzymes heattreatment is always considered as an option at an early stage in their purification. This method has been particularly successfully applied to the production of glucose isomerase, where a short incubation at a relatively high temperature is used (e.g. 60 - 25C for 10 min). No interfering activity remains after this treatment and the heat-treated, and hence leaky, cells may be immobilised and used directly. 1. Enzyme purification by Chromatography For identification and separation of biochemical compound chromatography is one of the most effective techniques. The method was developed in 1906 by the Russian botanist Mikhail Tswett. Chromatography involves the principle of partition or distribution coefficient ( Kd ). Kd describes that how a substance distributes itself between different type of immiscible phases. Enzyme preparations that have been clarified and concentrated are now in a suitable state for further purification by chromatography. For enzyme purification there are three principal types of chromatography utilizing the ion-exchange, affinity and gel exclusion properties of the enzyme, usually in that order. Ionexchange and affinity chromatographic methods can both rapidly handle large quantities of crude enzyme but ion-exchange materials are generally cheaper and, therefore, preferred at an earlier stage in the purification where the scale of
operation is somewhat greater. Gel exclusion chromatography (also sometimes called 'gel filtration' or just 'gel chromatography' although it does not separate by a filtering mechanism, larger molecules passing more rapidly through the matrix than smaller molecules) is relatively slow and has the least capacity and resolution. It is generally left until last as an important final purification step and also as a method of changing the solution buffer before concentration, finishing and sale. Where sufficient information has been gathered regarding the size and variation of charge with pH of the required enzyme and its major contaminants, a rational purification scheme can be devised. A relatively quick analytical method for obtaining such data utilises a two-dimensional electrophoresis whereby electrophoresis occurs in one direction and a range of pH is produced in the other; movement in the electric field determined by the size and sign of a protein's charge, which both depend on the pH. As the sample is applied across the range of pH, this method produces titration curves (i.e. charge versus pH) for all proteins present. A large effort has been applied to the development of chromatographic matrices suitable for the separation of proteins. The main problem that has had to be overcome is that of ensuring the matrix has sufficiently large surface area available to molecules as large as proteins (i.e. they are macroporous) whilst remaining rigid and incompressible under rapid elution conditions. In addition, matrices must generally be hydrophilic and inert. Although the standard bead diameters of most of these matrices are non-uniform and fairly large (50 150 m), many are now supplied as uniform-sized small beads (e.g. 4 - 6m diameter) which allows their use in very efficient separation processes (high performance liquid chromatography, HPLC), but at exponentially increasing cost with decreasing bead size. Relatively high pressures are needed to operate such columns necessitating specialized equipment and considerable additional expense. They are used only for the small-scale production of expensive enzymes, where a high degree of purity is required (e.g. restriction endonucleases and therapeutic enzymes). Column manufacturers now supply equipment for monitoring and controlling chromatography systems so that it is possible to have automated apparatus which loads the sample, collects fractions and regenerates the column. Such equipment must, of course, have fail-safe devices to protect both column and product. a. Ion-exchange chromatography Enzymes possess a net charge in solution, dependent upon the pH and their structure and isoelectric point enzyme can be separated. Note that In solutions of pH below their isoelectric point amino acid will be positively charged and bind to cation exchangers whereas in solutions of pH above their isoelectric point amino acid will be negatively charged and can bind to anion exchangers. Therefore pH chosen must be sufficient to maintain a high, but opposite, charge on both protein and ion-exchanger and the ionic strength must be sufficient to maintain the solubility of the protein without the salt being able to successfully compete with the protein for ion-exchange sites. The binding is predominantly reversible and its strength is determined by the pH and ionic strength of the solution and the structures of the enzyme and ion-exchanger. Normally the pH is kept constant and enzymes are eluted by increasing the solution ionic strength. A very wide range of ion-exchange resins, cellulose derivatives and large-pore gels are available for chromatographic use. Ion-exchange materials are generally water insoluble polymers containing cationic or anionic groups. Cation exchange matrices have anionic functional groups such as -SO3-, -OPO3- and -COO- and anion exchange matrices usually contain the cationic tertiary and quaternary ammonium groups, with general formulae -NHR2+ and -NR3+. Proteins become bound by exchange with the associated counter-ions. Ion-exchange polystyrene resins are eminently suitable for large-scale chromatographic use but have low capacities for proteins due to their small pore size. Binding is often strong, due to the resin hydrophobicity, and the conditions needed to elute proteins are generally severe and may be denaturing. Nevertheless such resins are a potential means of concentrating or purifying enzymes.
Ion-exchange cellulose and large pore gels are much more generally suitable for enzyme purification and, indeed, many were designed for that task. A variety of charged groups, anionic or cationic, may be introduced. The practical level of substitution of cellulose is limited as derivatisation above one mole per kilogram may lead to dissolution of the cellulose. Consequently, proteins may be eluted from them under mild conditions. Ion-exchange cellulose can be used in both batch and column processes but on a large scale they are used mainly batch wise. This is because the increased speed of large-scale batch wise processing and the avoidance of the deep-bed filtering characteristics of columns outweigh any advantage due to the increase in resolution on columns. Careful preparation before use and regeneration after use is essential for their effective use. Batch wise operations involve stirring the pretreated and equilibrated ionexchanger with the enzyme solution in a suitable cooled vessel. Adsorption to the exchangers is usually rapid (e.g. less than 30 minutes) but some proteins can take far longer to adsorb completely. Stirring is essential but care must be taken not to generate fine particles (fines). Unadsorbed material may be removed in a variety of manners. Basket centrifuges are a particularly convenient means of hastening the removal of the initial supernatant and the elution of the adsorbed material. This is usually done using stepwise increases in ionic strength and/or changes in pH but it is possible to place the exchangers, plus adsorbed material, in a column and elute using a suitable gradient. However, whilst ion-exchange cellulose are widely used for column chromatography on the laboratory scale, their compressibility causes difficulty when attempts are made to use large scale columns. Some of the problems with derivatised cellulose may be overcome using more recently introduced materials. Derivatives of cross-linked agarose (Sepharose CL6B) and of the synthetic polymer Trisacryl have high capacities (up to 150 mg protein ml-1) yet are not significantly compressible. In addition, they do not change volume with pH and ionic strength which allows them to be regenerated without removal from the chromatographic column. More detail description is given in chapter 9. 2 Affinity chromatography This is a term which is based on specific interaction between the enzyme and the immobilised ligand. In its most specific form, the immobilised ligand is a substrate or competitive inhibitor of the enzyme. Ideally it should be possible to purify an enzyme from a complex mixture in a single step and, indeed, purification factors of up to several thousand-fold has been achieved. An alternative, equally specific approach is to use an antibody (they are specific in binding because they are raised against specific antigen inside an animal) to the enzyme as the ligand. Such specific matrices, though, are very expensive and cannot be generally employed on a large scale. Additionally, they often do not perform as well as might be expected due to non-specific binding effects. In general, affinity chromatography achieves a higher purification factor (with a median value in reported purifications of about ten fold) than ion-exchange chromatography (with a median performance of about three fold), in spite of it generally being used at a later stage in the purification when there is less purification possible. A less specific approach, suitable for many enzymes, is to use analogues of coenzymes, such as NAD+, as the ligand. This method has been used successfully but has now been superceded by the employment of a series of water soluble dye as ligand. These are much cheaper and, usually by trial and error, have been found to have surprising degrees of specificity for a wide range of enzymes. This dyeaffinity chromatography was allegedly discovered by accident, certain enzymes being found to bind to the blue-dyed dextran used, as a molecular weight standard, to calibrate gel exclusion columns. More detail description is given in chapter 9. .3 Hydrophobic interaction chromatography (HIC) or affinity elution HIC is found to be very useful when it was noted that certain proteins were unexpectedly retained on affinity columns containing hydrophobic spacer arms.
Hydrophobic adsorbents now available include octyl or phenyl groups. Hydrophobic interactions are strong at high solution ionic strength so samples need not be desalted before application to the adsorbent. Elution is achieved by changing the pH or ionic strength or by modifying the dielectric constant of the eluent using, for instance, ethanediol. A recent introduction is cellulose derivatised to introduce even more hydroxyl groups. This material (Whatman HB1) is designed to interact with proteins by hydrogen bonding. Samples are applied to the matrix in a concentrated (over 50% saturated, > 2M) solution of ammonium sulphate. Proteins are eluted by diluting the ammonium sulphate. This introduces more water which competes with protein for the hydrogen bonding sites. The selectivity of both of these methods is similar to that of fractional precipitation using ammonium sulphate but their resolution may be somewhat improved by their use in chromatographic columns rather than batch wise. CHOICE OF MATRIX Careful choice of matrices for affinity chromatography is necessary. Particles should retain good flow and porosity properties after attachment of the ligand and should not be capable of the non-specific adsorption of proteins. Agarose beads fulfill these criteria and are readily available as ligand supports . Affinity chromatography is not used extensively in the large-scale manufacture of enzymes, primarily because of cost. Doubtless as the relative costs of materials are lowered, and experience in handling these materials is gained, enzyme manufacturers will make increased use of these very powerful techniques. Isoelectric focusing When any enzyme mixture is placed in the gel having solution of different pH then after passing the current a potential difference is established which helps in development of a pH gradient. This pH gradient decides the final movement of the enzyme in the gel, because enzyme will stop its movement only after a situation where pH of enzyme will be equal to the pI of the buffer. (See figure below, enzyme A 1 forming separate bands in compare to the Enzyme A2). Electrophoresis This method was developed by Arne Tiselius in 1937 and is based on principle that any charged molecule will move towards the opposite pole. More detail account is given in chapter 9. 2.11 Maintaining Enzyme Activity The key to maintaining enzyme activity is maintenance of conformation, so preventing unfolding, aggregation and changes in the covalent structure. Three approaches are possible: 1. use of additives, 2. the controlled use of covalent modification, and 3. Enzyme immobilization. In general, proteins are stabilized by increasing their concentration and the ionic strength of their environment. Neutral salts compete with proteins for water and bind to charged groups or dipoles. This may result in the interactions between an enzyme's hydrophobic areas being strengthened causing the enzyme molecules to compress and making them more resistant to thermal unfolding reactions. Not all salts are equally effective in stabilizing hydrophobic interactions, some are much more effective at their destabilization by binding to them and disrupting the localized structure of water (the chaotropic effect, Table 2.4). From this it can be seen why ammonium sulphate and potassium hydrogen phosphate are a powerful enzyme stabilizers whereas sodium thiosulphate and calcium chloride destabilize enzymes. Many enzymes are specifically stabilized by low concentrations of cations which may or may not form part of the active site, for example Ca2+ stabilises amylases and Co2+ stabilises glucose isomerases. At high concentrations (e.g. 20% NaCl), salt discourages microbial growth due to its. osmotic effect. In addition ions can offer some protection against oxidation to groups such as thiols by salting-out the dissolved oxygen from solution. Table 2.4. Effect of ions on enzyme stabilization. increased chaotropic effect
Cations Al3+, Ca2+, Mg2+, Li+, Na+, K+, NH4+, (CH3)4N+ Anions SCN-, I-, ClO4-, Br-, Cl-, SO42-, HPO42-, citrate3increased stabilization
________________________________________ Low molecular weight polyols (e.g. glycerol, sorbitol and mannitol) are also useful for stabilizing enzymes, by repressing microbial growth, due to the reduction in the water activity, and by the formation of protective shells which prevent unfolding processes. Glycerol may be used to protect enzymes against denaturation due to ice-crystal formation at sub-zero temperatures. Some hydrophilic polymers (e.g. polyvinyl alcohol, polyvinylpyrrolidone and hydroxypropylcelluloses) stabilise enzymes by a process of compartmentalisation whereby the enzyme-enzyme and enzyme-water interactions are somewhat replaced by less potentially denaturing enzyme-polymer interactions. They may also act by stabilizing the hydrophobic effect within the enzymes. Many specific chemical modifications of amino acid side chains are possible which may (or, more commonly, may not) result in stabilization. A useful example of this is the derivatisation of lysine side chains in proteases with N-carboxyamino acid anhydrides. These form polyaminoacylated enzymes with various degrees of substitution and length of amide-linked side chains. This derivatisation is sufficient to disguise the proteinaceous nature of the protease and prevent autolysis. Important lessons about the molecular basis of thermostability have been learned by comparison of enzymes from mesophilic and thermophilic organisms. A frequently found difference is the increase in the proportion of arginine residues at the expense of lysine and histidine residues. This may be possibly explained by noting that arginine is bidentate and has a higher pKa than lysine or histidine (see Table 2.1). Consequently, it forms stronger salt links with bidentate aspartate and glutamate side chains, resulting in more rigid structures. This observation, among others, has given hope that site-specific mutagenesis may lead to enzymes with significantly improved stability . In the meantime it remains possible to convert lysine residues to arginine-like groups by reaction with activated urea. It should be noted that enzymes stabilised by making them more rigid usually show lower activity (i.e. Vmax) than the 'natural' enzyme. Enzymes are more stable in the dry state than in solution. Solid enzyme preparations sometimes consist of freeze-dried protein. More usually they are bulked out with inert materials such as starch, lactose, carboxymethylcellulose and other poly-electrolytes which protect the enzyme during a cheaper spray-drying stage. Other materials which are added to enzymes before sale may consist of substrates, thiols to create a reducing environment, antibiotics, benzoic acid esters as preservatives for liquid enzyme preparations, inhibitors of contaminating enzyme activities and chelating agents. Additives of these types must, of course, be compatible with the final use of the enzyme's product.
Chapter-12 Techniques used in Enzyme characterization 2.12 HOW TO KNOW THE PROPERTY OF ENZYME 1. Calculate the molecular weight by following method Ultracentrifugation. Gel filtration.
SDS page. Mass spectrometry. 2. Presence of amide or peptide bond can be calculated by UV-IR spectroscopy. Direct structure can be determined by the X-ray crystallography. Amino acid can be determined by Ninhydrin reaction and taking OD at 570 nm. Amino acid sequencing can be done to know the order of amino acid. Introduction After salting in and salting out procedure, enzymes are further purified by two main common techniques 1- Chromatography; 2- Gel electrophoresis. In this chapter we will focus mainly on choice of these techniques and their broad details of the application and principles. A. Chromatography Principles of chromatography For identification and separation of biochemical compound chromatography is one of the most effective techniques. The method was developed in 1906 by the Russian botanist Mikhail Tswett. Chromatography involves the principle of partition or distribution coefficient (Kd). Kd describes that how a substance distributes itself between different type of immiscible phases. Basically, there is several type of chromatographic technique based on following property Adsorption. 2-Ion-exchange; 3-Molecular-sieve Adsorption Chromatography The stationary phase in adsorption chromatography is silica or alumina particles. Analyte are separated due to their varying degree of adsorption onto the solid surfaces. The main advantage of adsorption chromatography is in separating isomers, which can have very different physisorption characteristics due to steric effects in the molecules. Example Thin layer chromatography. Many specialized technique that use them are: --Basically, all type of chromatography consists of two type of phase Stationary phase: the phase that can not move and is prepared by dissolving some solid , liquid or other material that can be packed inside the column and may provide the support for movement the mobile phase. This may be solid, gel, liquid, or solid liquid mixture. Stationary phase in paper chromatography is the paper, while in ion-exchange chromatography is the ion-exchanger like Dowex -50 , Dowex-1 both are anion exchanger, and the cation exchanger is the CMC carboxymethyl cellulose, DEAE and sephadex. Mobile phase: This is chosen according to the nature of the biomolecules. Mobile phase is used to isolate the biomolecules because most of the biomolecules are present in the solution. Mobile phase is poured in the gel and get separated due to attachment with the stationary phase. The basis of all type of chromatography is the partition or distribution coefficient (Kd). Two immiscible phases are formed after the distribution of compound in the matrix and is denoted by Kd= concentration of compound in phase A / concentration of compound in phase B. 1. Column chromatography is of following type a) GPC or gel permeation chromatography b) Ion Exchange chromatography. c) Affinity chromatography. d) High performance liquid chromatography (HPLC) Based on adsorption of solute in the matrix, adsorption chromatography are of following type
a. Thin layer chromatography. b. Paper chromatography. COLUMN CHROMATOGRAPHY In the column chromatography column is filled with the stationary phase attached to suitable matrix and mobile phase passed through the column either with the gravity or by applied pressure. Size exclusion chromatography/ gel filtration (permeation) chromatography History Since the discovery in the 1940s that certain porous material can retain molecules of certain sizes and let others elute, there have been many discoveries that have made size exclusion chromatography practical. Size exclusion chromatography was discovered using starch as a packing material. Although starch was not a very durable material, it led to further interest. Soon, a material called Sephadex, developed for zone electrophoresis, was applied to chromatography; this innovation made size exclusion chromatography realistic. Sephadex is a cross-linked styrene divinylbenzene copolymer, which has led to the development of other divinylbenzene polymers for size exclusion chromatography. The amount of cross-linking (controlled by the availability of divinylbenzene) determines pore size, allowing packing materials to be easily customized for different applications. This initial use of size exclusion was limited to aqueous solvents and called gel filtration chromatography (at Tiseliuss suggestion). The process developed using Sephadex was used extensively in characterizing polymers and polymer molecular weight distributions. When using nonpolar organic solvents; researchers prefer modified divinylbenzene and polyacrylamide as packing materials. Silica has also been used, although the surface is often modified by adding organic compounds to resist adsorption. Today, both gel filtration and gel permeation chromatography are generally referred to as size exclusion chromatography; the processes are still used extensively in polymer chemistry but have gained new uses in biochemistry. Principle Size Exclusion Chromatography differs from conventional chromatography in the behavior of the stationary phase. In most forms of chromatography, the stationary phase chemically interacts through adsorption, charge, or various other actions. In size exclusion chromatography, the stationary phase simply acts as a sieve to filter molecules based on size. Term gel filtration or exclusion or permeation chromatography is used to describe the separation of molecule of different molecular size utilizing gel material, using molecular sieve. Here separation of molecule is done on the basis of their molecular size and shape with the help of gel that have pores formed during the gel formation. The larger size molecule are separated out or excluded from the pores (see figure), but still they can pass though the spaces around the pore called as void space and appear in the effluent first. Smaller molecules will be distributed between the mobile phases inside and outside the molecular sieve. They will pass through the column at slower rate. Hence they appear last in the effluent. Note that in the column there is always an inert substance. This is the reason why molecules are eluted from the column in order of decreasing size or if the shape is relatively constant. There are various materials that are porous in nature. The most commonly used material is organic gel and they form different type of pores on polymerizations. Therefore, the General principle is simple. The gel is filled in the column that is equilibrium with the mobile phase. Since gel have pores after polymerization and it can be used for the separation of the molecule of fixed size. Kd, denotes the distribution of an analyte (that we have to separate) in a column of a gel is determined solely by the total mobile phase, both inside and outside the gel particle. For given type of gel Kd depends on the molecular size of the analyte. If the analyte is large then kd =0, whereas if the analyte is sufficiently small to gain complete access to the inner mobile phase. So Kd =1. According to availability of the mobile phase kd varies between 0 to 1.
For two substances of different relative molecular mass and kd value for example Kd and kd is the difference in their elution volumes, Vs, can be derived from the equation and shown to be: VS = (Kd- Kd) V1------------------------------------1
The resolution depends on the beads. The coarser beads are unable to hold the fluid, so poorer resolution results. For maximum resolution, superfine beads are used. The gel is a three dimensional network whose structure is usually random. The gels acts as molecular sieves consist of cross linked polymers that are generally inert, do not bind or react with the material that is being analyzed, and are uncharged. The space within the gel is filled with liquid occupies most of the gel volume. The gels currently in use are of three types: ---dextran, agarose, and polyacrylamide. They are used in the aqueous solution.Dextran is a polysaccharide composed of glucose residues. It is commercially available in the trade name Sephadex. They can not be used for molecules that have size more than 600, 000. the alkyl dextran is called as N, N- methylene bis acrylamide. They made strong beads. The product is called as Sephacryl S-300.Agarose is a linear polymer of Dgalactose. It forms a gel held together with crosslink of H-bonds. The pore size is usually much larger than sephadex. This is the reason why they can be used for the separation of the large macromolecular substance like protein, DNA.Polyacrylamide gels are produced by cross-linking acrylamide and N, Nmethylene-bis acrylamide. It is marketed in the name of Bio-Gel P. Mixed gel of polyacrylamide and agarose is known as Ultra-Gel. Beads can be prepared with defined degrees of porosity, average radii, and mean radii. In general, the porosity of the beads determines the size range of molecules that can be effectively separated- the fractionation range. The radii of the beads are more important for determining the capacity of the column to separate molecules of similar size. Gel permeation chromatography is normally considered to be a medium- to high resolution chromatographic (polishing) technique. Mechanism of separation by size in gel permeation chromatography For our discussion of the theory of gel permeation chromatography, we will make the assumption that many proteins have a roughly spherical shape (often called globular proteins, as opposed to fiber proteins). Access to included volume (volume that hydrates the inside of the bed) depends on the size of the sphere (the hydrodynamic radius) that each protein occupies. The pore size of the bead will determine whether a protein of s Moreover, since proteins have roughly constant density (mass per volume), the size of this sphere is directly related to the mass of the globular protein. Thus, gel permeation chromatography is found to separate globular proteins according to mass. Note, however, that large deviations from spherical shape can cause a protein to migrate anomalously during gel permeation chromatography that is with regard to migration versus mass. Void volume, etc. For chromatography beads of a given porosity, molecules above a certain size will be completely excluded from the pore structure of the beads. These excluded molecules elute in the "void volume" (Vo) of the column and are not fractionated. In contrast, molecules below a certain size for beads of a given porosity will completely penetrate the pore structure and will elute with the
"included volume" (Vi). Thus, these molecules are not fractionated either. Only molecules with masses (hydrodynamic radii) that allow them to penetrate the pore structure of the beads only partially fall within the fractionation range and are separated according to their molecular size (approximately, as described above).The use of a gel permeation column as an analytical tool requires the determination of Vo, Vi, and the elution volumes (Ve) of the proteins of interest. By comparing the elution volumes of protein standards of known molecular mass, the molecular mass of an unknown protein can be estimated. In contrast to denaturing gel electrophoresis, which gives subunit molecular masses for an oligomeric protein, gel permeation chromatography provides an estimate of mass for the oligomeric holoprotein. The combination of denaturing gel electrophoresis and gel permeation chromatography is thus exceedingly powerful for elucidating basic features of protein structures. Use of gel permeation chromatography as part of a purification scheme Most protein purification schemes involve several steps. These can be characterized as either bulk or polishing steps. Bulk purification steps are generally useful for samples of low purity and large volume, both of which are typical of the starting material to be used in a protein purification, such as a cellular extract. Common bulk purification methods are centrifugation; precipitation, such as with salt, solvent, acid, or polyethylene glycol; filtration; and ion-exchange chromatography. Polishing purification steps are generally useful for samples of higher quality and smaller volume, typical of partially purified fractions obtained from bulk purification steps. Common polishing purification methods are ion exchange, affinity, and gel permeation chromatography. Gel permeation chromatography is a common and often excellent polishing step during any protein purification; however, it would not be used as a first step in purification except in unusual circumstances (e.g., high abundance of the target protein in the starting material). Table:1 MATERIAL: POLYMER 1.DEXTRAN < 0.7 SEPHADEX 1.0-5 1.53.0 4--150 5---600 SEPHYCRYL 5---250 10---1500 208000 2. AGAROSE 10-4000 SEPHAROSE 60---20,000 4B 2 B G S 200 200 S 300 G 50 G 100
TRADE NAME
FRACTIONATION G 10 G 25
RANGE
S 400
6B 7040,000 A 5m 10---5000 15,000 100---50,000 3. POLYACRYLAMIDE 0.1---1.8 1.06 BIO-GEL P2 P6 BIO-GEL A 50 A 15m 40---
P100 5.0--100.0 Overview of a typical gel permeation chromatography experiment In a typical gel permeation experiment, a protein sample (often a partially purified fraction from a previous purification step, see below) is applied onto the top of the column packing (beads) and allowed to flow into them. As additional buffer is applied to the top of the column and allowed to flow through the column, the protein sample migrates through the column. Due to the differential partitioning of molecules into the pore structure of the beads (as described above), the protein molecules are separated. As buffer is eluted from the column, it is collected in fractions until all the components of the original sample have passed through the column.
Note that all the large Ms come out at nearly the same volume, Vo. This is because none of the very large polymers ever enter a pore. So they all elute together at the void volume, Vo. It is customary to plot log(M), not M. Note that the independent variable is plotted on the y axis by convention. With such a curve, you are to select a representative sampling of points. For
example, consider point A, you hit the M vs. Ve trend Obtain DRIA similarly from wish! The DRI response is DRI c ( in g/mL)
indicated by the cross. Starting at VeA read up until and then read left to get MA from the left y-axis. the right ordinate. Repeat for as many points as you proportional to the concentration of polymer:
The constant of proportionality is dn/dc, the same specific refractive index increment needed in light scattering. You cannot measure concentrations in an isorefractive solvent (i.e., one in which dn/dc = 0). However, it is not necessary to actually know dn/dc in simple GPC. One can obtain average molecular weights without it. For example: Mw = =
Since, the constant of proportionality factors out of the numerator and denominator identically. One can also obtain the number average molecular weight: Mn = = = =
ADVANTAGE OF GEL CHROMATOGRAPHY In this chromatography the material used is inert one so there is no effect of temp. pH, ionic strength. They can be used for very labile material like enzyme. Since there is no adsorption by the inert material so labile material are not affected. The elution volume is related in a simple manner to molecular weight. Application of GPC or gel permeation chromatography Size exclusion chromatography is used primarily in two areas of chemistry: polymer chemistry and biochemistry. In polymer chemistry, size exclusion chromatography can separate polymers with different numbers of monomer units, giving the polymers producer information about the length of the chains produced. In addition, because size exclusion chromatography does not destroy or alter the sample, the fractions of polymer can be further tested. For example, S. J. ODonohue and E. Meehan discuss the use of different solvents in size exclusion chromatography and molecular weight detectors (light scattering and viscometry) at high temperatures to characterize polymers, such as o-chloronaphthalene, that can be processed only at temperatures above 135 C (1999). Antonio Moroni and Trevor Havard discuss the use of solvents in the analysis of engineering thermoplastics, particularly polyesters and polyamides (1999). Their experimentation leads to the suggestion that 1,1,1,3,3,3-hexafluoro-2-propanol may not be a good solvent for size exclusion chromatography as polar portions of macromolecules (such as polyesters and polyamides) may be solvated and extended, making these molecules seem larger than they actually are. This extension or enlargement causes them to be eluded at later times than expected. In biochemistry, size exclusion chromatography is useful in separating highmolecular-weight molecules from other molecules. For example, in a genetics study, Anke van Rijk and colleagues used size exclusion chromatography to isolate a small protein called alpha-A-crystalline to study the insertion of hamster alpha-Acrystalline into mice (1999). size exclusion chromatography allows the researchers to isolate the biomacromolecule based on its relative size without destroying the sample. Biochemical separations may seem to make this more of a separation technique, but in both polymer chemistry and biochemistry, size and molecular weight estimations can be made. If a polymer chain of unknown size, or a protein of unknown size, is
analyzed along with molecules of known size or molecular weight, then when the compound in question eludes, it can be compared with the molecules that it eluded closest to determine an approximate molecular weight. In purification of Macromolecule: GPC can be used to separate the viruses, proteins, hormones, enzyme, antibodies, nucleic acid and polysaccharide. In study of protein binding study Estimation of the molecular weight: Gel 1 gel 2 Log M THE plot is between K verses log M (molecular weight). It yields a straight line except for very small and very large molecule. The parameter K = Ve---Vo / Vs, where Ve= elution volume, Vo= void volume, Vs= volume of stationary phase or = Vt---Vo, Vt is the total volume of the column PAPER CHROMATOGRAPHY Cellulose has many hydroxyl groups which are polar and bind H2O. The bound H2O is the Stationary Phase. H2O run across the cellulose paper due to capillary action. Mobile Phase is mixture of organic solvents (alcohols, ketone, aldehyde etc.) and possibly water. The mobile phase solvent will be less polar than H2O; it is usually a mixture of Solutes partition between H2O in the stationary phase and the solvent of the mobile phase. One can change the characteristics of separation by changing the polarity of the mobile phase (i.e. adjust the composition). Paper Chromatography is the most common form of cellulose chromatography in which solute is "spotted" on "dry" paper (still contains H2O) encircle by the pencil at a line mark and chromatograph is "developed by dipping one end in the mobile phase. There are two modes of chromatography1: Ascending and 2-Descending.The solvent moves through the paper, drawn by capillary action Solutes move as spots with a rate depending upon how much time they spend in the stationary phase vs. the mobile phase--determined by their partition coefficient--measured as an Rf value. Maximum distance covered by the solvent is noted and the distance covered by the solute is noted. The ratio of two gives Rf value. This Rf value is fixed and is generally less than one. Ion exchange chromatography This type of chromatography is used for many biological materials like amino cid and proteins, which have ionisable groups, i.e. they have either negative or positive charge. The name ion exchange is given because of exchanging ions for ion in aqueous solution. The principle of ion exchange chromatography is that charged molecule adsorb to ion exchangers reversibly so that molecule can be bound or eluted by changing the ionic environment. Separation by ion exchange is usually involving two step. First the substances to be separated are bound to the exchanger, using the conditions that make them more stable and tight binding. Ion exchange separation is carried out mainly in column packed with an ion exchangers. The column packing for ion chromatography consist of ion-exchange resins bonded to inert polymeric particles (typically 10 m diameter). For cation separation the cation-exchange resin is usually a sulfonic or carboxylic acid, and for anion separation the anion-exchange resin is usually a quaternary ammonium group. For cation-exchange with a sulfonic acid group the reaction is: -SO3- H+(s) + Mx+(aq) -SO3- Mx+(s) + H+(aq) where Mx+ is a cation of charge x, (s) indicates the solid or stationary phase, and (aq) indicates the aqueous or mobile phase. The equilibrium constant for this reaction is: [-SO3- Mx+]s [H+]aq Keq = ------------------------------------------
[-SO3- H+]s [Mx+]aq Different cations have different values of Keq and are therefore retained on the column for different lengths of time. The time at which a given cation elutes from the column can be controlled by adjusting the pH ([H+]aq). Most ion-chromatography instruments use two mobile phase reservoirs containing buffers of different pH, and a programmable pump that can change the pH of the mobile phase during the separation.
There are two type of ion exchanger 1) cationic exchanger 2) Anion exchanger. Cationic exchanger posses negative charge, while anionic exchanger posses positive charge. The negative charge ion exchangers bind to positive charge molecule. The more highly charged molecule to be exchanged, the tighter it binds to the exchanger and less readily it is displaced by other ion. A typical group used in ion exchange is the sulphonic group, SO3 -. If an H+ is bound to group, the exchanger is said to be in the acid form. It can exchange one H+ for one Na+ or two H+ for one Ca++. The sulphonic acid groups are called as a strongly acidic cation exchanger. Other weakly acidic cation exchangers are phenolic hydroxyl group and carboxylic group. Strong Ion Exchangers: based upon strong acids or bases and are charged over a wide range of pH a. strong cation exchangers: Sulfonic Acid (or derivatives) RSO3- b.strong anion exchangers: quaternary ammonium salts R-N (CH3)+ Weak Ion Exchangers: based upon weak acids or bases which are charged only over a limited pH range a. weak anion exchangers: Diethyl-amino-ethyl (DEAE); tertiary amines b.weak cation exchangers: carboxy methyl (CM); phosphoryl. These may be bonded to a variety of supports: e.g. DEAE-cellulose; DEAE-Sephadex; DEAE-Sepharose; CMCellulose; CM-Sephadex; CM-Sepharose Choosing an Ion Exchanger Charge -- cation or anion exchanger -- depends upon the charge of the molecules to be separated. This will also depend upon pH. Weak Exchanger is useful for labile molecules such as proteins. Strong Exchanger is used for more stable molecules such as nucleotides, amino acids, peptides etc.The matrix is used of various materials like dextran, agarose, cellulose and copolymer of styrene and vinyl benzene. The total capacity of ion exchanger is measure of its ability to take up exchangeable ion. It is measured in term of milliequivalent of exchangeable group permiligram of dry weight. The available capacity is the capacity under particular conditions like pH, ionic strength. The extent to which an ion exchanger is charged depends upon the pH. The porosity of the matrix is very important factor because of charged group are present both outside and inside of the matrix. Since pore also act like molecular sieve. Large molecules may be unable to penetrate the pore so the capacity will decrease with increase in molecular weight. Ion exchanger come in variety of particle size called as mesh size. Finer mesh means an increased surface to volume ratio and therefore increased capacity and decrease time for exchange to occur for given volume of exchanger. Finer the mesh the slower will be the flow rate. For materials that have either single charge the choice of exchanger is not difficult. But in case of material that have both negative and positive charge the choice depends upon the charge that is stable. As we know above the isoelectric point if the pH is stable one then an ion exchanger can be used. If stable below isoelectric point then a cation exchanger can be used. If the substance is labile, a weak exchanger can be used. The sephadex and bio-gel exchanger offers a particular advantage for macromolecules that are unstable in low ionic strength. Because the cross links in these materials maintain the insolubility of the matrix even cross link in these materials maintain the insolubility of the matrix even if the matrix is highly polar.
Macromolecular separation always needs larger pore size while micro molecule can be separated by the small pore. It is because of larger available capacity. The cellulose ion exchanger proved to be best for the macromolecule like protein and the nucleic acid. Small mesh sizes improve the resolution but decrease flow rate, which increases the zone spreading and decrease resolution. For selection of buffer following rule is always adopted. The cationic buffer is used with the anionic exchanger, and anionic buffer is used with the cationic exchanger. Simply because, the binding strength, depends on the ionic factor. For best ionic resolution ionic condition should be same as the eluting condition. Eluting Ion Exchange Columns -- molecules usually adsorb tightly in the column so, this interaction, must be weakened. (Most common method): F = q1 q2 / D r2 q1and q2 are the charges on two groups, r is the distance between the groups, and D is the dielectric constant of the solvent which is increased with higher ionic strength thus weakening the force between the solute and the ion exchanger. Another way to look at this is that other ions in the buffer compete for the ion exchanger binding site.change pH -- changes the charges on the molecules being separated; also can change the charge of a weak ion exchanger .These changes can be made stepwise by changing the buffer reservoir (step gradient) or as gradient -- by mixing two buffers The basic principle of ion exchange chromatography is that the affinity of substance for the exchanger depends on both the electrical properties of the material and the relative affinity of other charged substances in the solvent. Hence, bound material can be eluted by changing pH, thus altering the charge of the material. In deciding the eluting condition, it is important to consider how eluting fluid will affect the assay for the material. For example if spectral analysis is to be done then eluting fluid should not absorb in the required wavelength. One important use of on exchange is in desalting. Gel chromatography is an effective means of removing ion from solutions of macromolecules. A related resin called a mixed bed resin is used to prepare deionized water. Ion exchanger has been found to be an effective way to separate weakly polar substances by using the exchanger as the matrix for partition chromatography. For example to separate the sugar mixed with the polar and nonpolar solvent, the solution is applied to the column. The polar solvent binds to the matrix forming the polar stationary phase and sugar partition between this phase and the mobile weakly phase. This is called as reverse phase chromatography. Gas / Liquid Chromatography In gas chromatography mobile Phase is usually a gas -- usually inert (He, Ar, N2) and Stationary Phase is a liquid coating on an inert solid support. There is open tube or capillary operation and other a coat the inside surface of a long thin tube (30 - 100 m long) Packed column -- larger diameter column is packed with an inert support -commonly diatomaceous earth, teflon powder, or glass beads . Coating may be solid at room temp (e.g. polyethylene glycol) but the column is run at higher temperature where the coating melts. Sample usually injected as a liquid which is then heated to vaporize it. Note that sample must be somewhat volatile and stable at higher temperature. GC Can provide very high resolution by making very long columns. Detectors present are of various types. Most powerful detector is a mass spectrometer (GC/Mass Spec.) which give mass spectrum that can be used to identify and quantitate samples as them come off the column. Reversed Phase Chromatography In reverse phase chromatography stationary Phase is apolar (hydrophobic) and is reversed with respect to cellulose chromatography. This hydrocarbon chains is bound to an inert matrix to provide hydrophobicity. Hydrophobicity can be varied by changing the hydrocarbon chain length or by aromatic groups. Other phase is mobile Phase that depends upon hydrophobicity of stationary phase. Commonly use a
more polar organic solvent like acetonitrile, DMSO, EtOH, ethylene glycol, propanol, or mixtures of these with H2O. Use of shorter hydrocarbon chains less densely packed is called Hydrophobic Interaction Chromatography HIGH PRESSURE LIQUID CHROMATOGRAPHY [HPLC] Conventional liquid chromatography uses plastic or glass columns that can range from a few centimeters to several meters. The most common lengths are 10-100 cm, with the longer columns finding use for preparative-scale separations. Highperformance liquid chromatography (HPLC) columns are stainless steel tubes, typically of 10-30 cm in length and 3-5 mm inner diameter. Short, fast analytical columns, and guard columns, which are placed before an analytical column to trap junk and extend the lifetime of the Stationary Phases. Usually the resolving power of a column increases with the length of the column and the number of theoretical plates per unit. The number of plates increases as the available surface area per unit length of the column becomes greater in other words as the matrix particle become smaller. However, the flow rate of a column drops as the particle size decreases and this allow sufficient time for significant diffusional spreading to occur. This decreases the resolution. Diffusional spreading can be reduced if the transit time of the mobile phase in the column made small. This can be done by establishing a pressure difference across the top and bottom of the column to force the liquid through the bed. The use of very fine particle and very high pressure to maintain adequate flow rate is called as HPLC or high performance or high pressure liquid chromatography. HPLC is known for its rapid separation with extraordinary resolution of peaks. Also it requires the less amount of test material. It is because of its small fraction of the void volume. For e.g. with a typical column having a diameter of 36mm and a length of 10-20 cm operating at 50-200 pounds per square inches [ 520 kg/ cm2 ] , a sample of 0.01 to 0.1 ml. can be used. The pressure used affects the resolution of the bands. At atmospheric pressure diffusion spreading causes the bands to overlap. However, if pressure (and hence flow rate is very high, there may be insufficient time for molecules in the mobile phase equilibrate and the bands are also broadened. At present time this procedure is applied principally with the ion-exchange and adsorption chromatography of small molecule, peptide, small carbohydrate and t-RNA.
Partition Chromatography In partition chromatography the stationary phase is bonded to inert particles of 3-10 m of diameter, with the smaller sizes, 3-5 m, being used in analytical columns, and the larger particles being used in preparative-scale HPLC. Analyte separate as they travel through the column due to the differences in their partitioning between the mobile phase and the stationary phase. Reverse-phase partition chromatography uses a relatively nonpolar stationary phase and a polar mobile phase, such as methanol, acetonitrile, water, or mixtures of these solvents. The most common bonded phases are n-octyldecyl (C18) and n-decyl (C8) chains, and phenyl groups. Reverse-phase chromatography is the most common form of liquid chromatography, primarily due to the wide range on analyte that can dissolve in the mobile phase. Normal-phase partition chromatography uses a polar stationary phase and a nonpolar organic solvent, such as n-hexane, methylene chloride, or chloroform, as the mobile phase. The stationary phase is a bonded siloxane with a polar functional group. The most common functional groups in order of increasing polarity are: cyano: -C2H4CN diol: -C3H6OCH2CHOHCH2OH amino: -C3H6NH2 dimethylamino: -C3H6N(CH3)2
Chromatographic Separation principle Commercial name Phase Type of support Adsorption Partisil C8 Corasil Pellumina Partisil Micro Pak Al Bondapak C18 ULTRA pak TSK ODS Octylsilane Silica Alumina Silica Alumina Porous Pellicular Pellicular Microporous Microporous Pellicular Porous Ion exchange Partisil-SAX MicroPak-NH2 Strong base Weak base Porous Porous Exclusion Styragel Bio-glass
Nature of stationary
Superpose Fractogel TSK Glass Polystyrene-divenyl benzene Agarose Polyvinylchloride Rigid solid Semi-rigid gel Soft-gel Semi-rigid gel Affinity chromatography Principle Affinity chromatography is almost exclusively used for the purification of biological molecules such as proteins and other macromolecules. The technique has been known for almost a century, but suitable support materials were not available until much more recently. With new materials and new demands, the technique became extremely useful in the 1960s, and it has become essential with the growing demand of the biotech in the 1980s and 1990s.Biotech research has used affinity chromatography because of its ability to separate one desired species from a host of other biological molecules. Specificity based on three aspects of affinitythe matrix, the ligand, and the attachment of the ligand to the matrixis the hallmark of this process and the reason for its success. As the name suggests, it utilizes the property of biological affinity of the substances to be separated. As a consequence, it is capable of giving absolute purification, even from complex mixtures, in a single process.
Affinity chromatography operates on the principle that ligand (as stated in the Table), attached to a matrix made up of an inert substance, bind to the desired molecule within a solution to be analyzed . Ideally, the ligand will interact only with the desired molecule and form a permanent bond. All other compounds in the solution will elude, leaving the desired product in the column. The desired molecule is then removed from the column by using a wash (typically changing the pH) that lowers the dissociation constant and allows recovery of a nearly pure sample. Choosing the correct ligand is the first hurdle. The ligand must bind strongly with the molecule that is to be recovered. Biological systems have millions of ligand (also known as receptors), and most can be affixed to the matrix and used to isolate the desired molecule. If the ligand chosen can bind to more than one molecule in the sample in question, then a technique called negative affinity, which uses ligand to remove everything but the target molecule from the solution, may be used. For example, if you were looking for a molecule from a cell, but the only ligand that binds the molecule also trapped two additional, different molecules, you could run a normal affinity column and collect these three molecules. Then, if a ligand were found that attached to the two unwanted molecules, that ligand could be used in the size exclusion chromatography and affinity column, allowing the desired molecule to elute while the other two were retained in the column. Matrix materials simply hold the active ligand and provide a pore structure to increase the surface area to which the molecules can bind. Ligand attachment requires that the matrix be activated and then react with the ligand to fix them onto the matrix. During this process, the ligand must also remain active toward the target molecule, or all will be for naught. Substituent groups within the matrix, such as amino, hydroxyl, carbonyl, and thio groups, are easily activated and can serve as the sites to which the ligand attach. Matrix materials are often polysaccharides, such as agarose, that have many hydroxyl groups that can be activated. The matrix, in addition to requiring activation, must also often stand up to decontamination when purifying pharmaceutical compounds. Decontamination is typically performed by rinsing the column with sodium hydroxide or urea. Different matrix materials are stable in different pH ranges, adding the third aspect of selection for affinity chromatography. The technique was originally developed for the purification of enzymes, but it has been extended to nucleotides, nucleic acids, immunoglobulin, and membrane receptors and even to whole cells and cell fragments. The technique can be shown by the following equation: if we assume M is the macromolecule and L ligand is attached to the matrix, then an ML complex will be formed. M + L ML
For the success of the experiment following steps must be kept in mind. 1. Matrix should have broad range of thermal and chemical stability. It should not absorb any chemical or substance to be purified itself. 2. The ligand should be coupled without altering its binding properties. 3. A ligand must bind tightly. 4. During the elution the matrix should not be destroyed. The most useful material is the Agarose, and polyacrylamide, since they have broad range of thermal tolerance, exhibit minimum absorption, maintain good flow property after coupling, and does not denature after application of extreme pH and ionic strength. The choice of ligand depends on the specificity of the substance. For example for
an enzyme ligand must be the substrate, a reversible inhibitor, or an allosteric activator. METHOD OF LIGAND IMMOBILIZATION Linking or coupling of the ligand to matrix material is called as immobilization. 1. CNBR activated agarose CNBr is negatively charged so they can react strongly with the amino group. It is extremely useful for coupling enzyme, Co-enzyme, inhibitors, antigen, antibody, nucleic acid and most protein to agarose. 2. 6-AMINO-HEXENOIC ACID & 1,6 DIAMINO HEXANE. a) Used mainly for small ligand. b) They can solve the steric problem c) Only a Spacer can be attached between matrix and ligand. d) Spacer can be attached by reacting functional group C00H (by using Agarose) and NH2 (group of 6AHA). e) Epoxy activated Agarose.This is useful for linkage of sugars and carbohydrate or any material containing OH gp, amino gp, thiol gp. f) Thiopropyl-Agarose.Can be Used or sulpher containing protein. Before using, it should be treated with the cysteine. 3. Carbomyl diimidazole activated agarose. Coupling of N-nucleophile to CNBr. This results in isourea linkage that carries potential charge and thus can act as an ion-exchanger. Table9. 2 Affinity chromatography Substance Use Lectin Polysaccharides and glycoprotein present on the RBC membrane. Con A Sepharose They selectively can binds to the N-acetyl glucosamine residue. Therefore they can be used for the lymphocyte separation. Helix pomatia lectin Used for separation of the pure T-cell Lactose Caster bean, pea nut can be used for separation of lactose. Maltose Can be separated by the jack bean. D-glucosamine Can be separated by clam NADP+ Can be separated by using 2, 5 ADP. Protein A Can be used for separation of IgG (using Fc region) Poly A Can be separated of m-RNA Boronate polyacrylamide RNA, sugar, catecholamine Heparin-Agarose Blood protein, DNA polymerase, Ribosome, androgen receptor Imino-diacetic acid Zn+2, Cu +2 Can be separated Octyl agarose Protein Can be separated Thio-propul Sepharose Can be separated by clam Protein, Urease, and Papain. Applications The applications of affinity chromatography have been numerous, but they are predominantly in the field of biochemistry. Early applications were in the separation of biomacromolecules from other biological compounds, such as molecules from an entire cell. This continues to be the primary and extremely important field for affinity chromatography. For example, S. Loukas and colleagues studied the existence of one type of suspected opiate receptor in the brain (1994). They separated the ( -opioid binding protein by using an opioid receptor antagonist that was specific to the ( -opioid binding protein. These studies may one day lead to a better understanding of how drugs such as opium affect our brains. In addition to using small ligand to separate large molecules, researchers have immobilized large molecules on the matrix and used them to separate the small molecules that bind to them. In addition to biological separation, affinity chromatography can be used to determine dissociation constants of ligand and molecules. The longer a molecule stays on the column, the broader the chromatographic peak, indicating that the molecule is more tightly bound to the ligand. This quantitative information can be used in further studies of the ligand
or molecule. Thin layer chromatography Principle in this type of chromatography the stationary phase is generally the glass plastic or metal foil plate. The mobile liquid phase passed through the thin layer plate. The layer is formed must be thin Mobile phase is passed through the stationary phase under capillary force, and the analyte is distributed well in the layer. The distribution coefficient is represented by the Kd. Distribution process is based on the fact of adsorption, partition, ion-exchange, exclusion chromatography. The movement of analyte is expressed by the retardation factor. Rf= origin. Distance moved by the solute from origin / Distance moved by the solvent from
K can be calculated by = 1Rf/ Rf Process of preparation of the thin layer plate A thin layer of about .25 mm thick slurry is prepared in water is applied to glass with the help of plate spreader. CaSO4 is mixed to facilitate adhesion of the adsorbent to the plate. The plate is dried at 1001200C. This is also helpful in the activation of the plate. Sample is applied to the plate 22.5 cm from edge by means of micropipette or microsyringe. The TLC plate is dipped in the developing phase to depth of about 1.5 cm it is left for one hour. Detection is done by the several methods. For e.g. fluorescent dye (this can be absorbed by the UV light. For investigating the unsaturated compound I2 vapour can be used. For radiolabel led compound autoradiography can be used. Movement of compound can be observed by the specific Rf values. ADVANTAGE OF TLC OVER OTHER CHROMATOGRAPHY TECHNIQUE IT has great resolving power and greater speed of separation. A wider range of sorbant can be used and also due to easy detection of spot and separation of chromatogram. Two factor that affect the TLC has, High surface to volume ratio and the weight ratio, If necessary 2D gels electrophoresis can be used in the TLC plate this can be used by placing the plate in the buffer at right angle to first separation. Commonly used separating agent is the Ninhydrin for detection of the amino acid. Rhodamine B is used for the detection of the lipid; SbCl2 for steroid and tarpenoid; H2SO4 for organic acid, H2SO4 and KMN03 for hydrocarbon. Br2 vapour is used for olefins detection. HPTLC and HPPLC Thin-layer chromatography (TLC) gets the high-performance (HP) treatment through several optimization steps leading to improved efficiency and automation. The layers are made smaller (0.02 mm instead of 0.25 mm), they have smaller mean grain sizes of the coating particles (down to 7 m instead of 1230 m), and the grain size is more uniform. Equally if not more important, HPTLC shows better optical properties than standard TLC, allowing for easy densitometry studies. Better resolution as well as a 10-fold improvement in detection limits are key to HPTLCs increasing popularity.
By using a forced-flow mobile phase and a sealing chamber, researchers can transform HPTLC into high-pressure planar liquid chromatography (HPPLC), gaining the added benefits that pressure provides, especially to the speed of the run. HPTLC can also be used with centrifugation to force the sample outward in what is known as rotational planar chromatography (RPC). These adaptations are collectively responsible for what is known as modern TLC, which relies heavily on instrumental analysis. Applications of these modern forms are extensive, including routine use in the pharmaceutical industry and clinical analysis, in food analysis, natural products analysis (including a variety of botanicals and herbals), and in environmental analysis for determining such things as pesticides in drinking water. Selection of chromatographic system i. Ligand having specific binding propertyAffinity chromatography. ii. Volatile compound ----gas liquid chromatography. iii. For compound having different functional groupadsorption chromatography. iv. Low polar compound---liquid chromatography. v. Water soluble but weakly ionic---reverse phase liquid chromatography. vi. Water soluble but strong ionic compound---ion-exchange chromatography. vii. Compound differ in molecular sizeexclusion chromatography. Electrophoretic technique Electrophoresis, migration due to an electric field, was first discussed by L. Michaelis (1909). Arne Tiselius rediscovered this technique in 1937 and used it to separate human serum proteins. In subsequent years, development focused on new gel materials that minimized convection, which interferes with charged species migration. In the 1960s, polyacrylamide gels were developed, which ushered in the widespread use of gels in biological studies. Capillary electrophoresis was pioneered by S. Hjerten, who built tube electrophoresis units as early as 1959.However, the technology on which this technique depends, such as high-quality capillary silica tubing and extremely sensitive detectors, had yet to mature. It took two more decades before Jorgen son and Lukacs developed the first truly useful CE instrument and showed that it had exceptional resolution (1981). Theory. CE uses the phenomenon of electroosmotic flow to separate analytes. CE instruments consist of a capillary column between two reservoirs of buffer solution (the solution used as the solvent). Electrodes are placed in both reservoirs, which places a potential across the column. The column itself becomes negative if the cathode is on the product side and the anode is on the sample side. When the column is negative, cations migrate toward the column wall, creating a dense +ve area. This area is then attracted by the cathode and starts to flow toward the product end of the column. These cations pull the rest of the fluid with them. A reversed-phase HPLC packing material is used to retain the analytes at different rates on the liquid stationary phase. Traditional CE uses polar solvents, precluding the identification and analysis of polar compounds. But in the early 1980s, Terabe and colleagues developed a variant of CE that allows determination of nonpolar molecules (1984, 1985). Micellar electrokinetic capillary chromatography (MEKC) uses surfactants or polymers to form micellessmall spheres with hydrophobic centers and ionic shields around the outsidethat travel against the liquid flow toward the anode (the charges could be switched with different surfactants) but are swept with the electroosmotic flow toward the cathode. However, they will elute last because of their struggle to reach the anode. Hydrophobic molecules will enter the micelle, but they will also exit in a fashion similar to liquid partition chromatography. Therefore, the separation of molecules is based on their polarity as polar molecules are slowed by entering the micelle. It describes the migration of a charged particle under the influence of electric field. Many important biomolecules can be separated by this technique like: amino acid, peptide, protein, nucleotide, ionisable group. The gel is prepared from the material like agarose or polyacrylamide and the separation of compound is done in the buffer. The substances separated at the low
temperature to save it from heating effect. The function of buffer is to maintain the constant ionization pattern of the molecule. Force can be represented by the following equation: F= qv/ velocity q-= charge and V is the potential difference. Electrophoretic mobility () is defined as ratio of velocity to field strength.
a. Agarose Gel Electrophoresis Agarose is prepared from natural compound isolated from Gracilaria and Geladium. The basic repeat unit is the galactose and 3,6- anhydro galactose. Generally the agarose concentration is used in the 1% to 3% . Agarose has property to dissolve in the hot water making liquid solution while it solidifies in the cooled medium and forms a rigid gel. The gel is formed due to inter and intra-H bonding. This cross linking attaches new anti conventional properties. The pore size is decided by the initial concentration of the agarose. High concentration of agarose gives usually smaller pore size. While, small concentration of agarose gives larger pore size. The main problems occurs during electrophoresis is the phenomenon of electro endosmosis. This can be avoided by the substituting some other functional group like sulphate. 1% Agar is used for the immuno-elctrophoresis or flat bed electrophoresis. Generally pore size of the 1% Agar has pore size larger than proteins and nucleic acid. Agar can be used more for analysis of DNA. For this purpose vertical slab gel can be used. b. Polyacrylamide Gel Electrophoresis After the polymerization of acrylamide it forms a polymer compound called as polyacrylamide. And so electrophoresis is called as polyacrylamide gel electrophoresis or PAGE. THERE is some necessary component for the polymerizations of the acrylamide like the N, N bis acrylamide. Note that bis acrylamide is essentially two acrylamide linked by the methylene group. Initiation of the polyacrylamide is difficult so some more compound is added like ammonium persulphate, and the TEMED or N,N,NN tetramethylenediamine. Actually addition of TEMED IS necessary as it acts like the catalyst. S2O8 + eSO4 2+ SO4
IF this free radical is represented as R. Oxygen can be used for the removal of the free radicals. Free radical generation can be done by the photo polymerization. Low percentage gel can be used for the separation of the DNA. For protein separation SDS PAGE can be used the usual percentage lies between the 3 to 30 % of acrylamide. Since protein separation done in the two steps: first it is separated in the stalking gel, and size exclusion chromatography on it is resolved by the resolving gel. Stalking gel generally uses lower percentage of the gel that provides the larger pore size. It allows the free movement of the protein so that one type of protein either same in the molecular weight or charge can stalk at one position. After this process the protein is separated in the gel having smaller pore size. All these process can be done by the vertical slabs. c. SDS-PAGE SDS is an anionic detergent (CH3(CH2)10 CH2OSO3-Na+). With mercaptoethanol and SDS, protein can be denatured into the simple protein. It helps in the better separation of the protein. And thus protein is analyzed in qualitative way. On average it has seen that one SDS can binds to the two amino acid. Problem arises during the electrophoresis is the settling of sample and exact tracking of the
protein position in the gel. For settling problem sucrose solution is used, and for tracking the protein bromo-phenol blue is used. The protein first of all loaded in the stalking gel. In this master mix, glycine is added. This is done to sharpen the protein band. But note that glycinate ion have a lower electrophoretic mobility than protein SDS complex, which have lower mobility than chloride ion of the loading buffer and the stacking gel Cl- > PROTEIN-SDS > GLYCINATE So protein SDS band lies in between the Cl- and glycinate ion. PH of the stacking gel is generally 6.8 and resolving gel has pH about 8.8. The negatively charged protein is attracted toward the anode. After the protein is reaches the bottom, the gel is removed and placed in stain more generally the Coomassie blue. The gel is then placed in the destain solution. Staining of gel is done for 2-3 hrs. And destaining requires overnight. Generally 15 % polyacrylamide gel is used in the separating gel. This can allow separation of the protein in the range of 100,000 to 10, 000. For protein of molecular weight more than 150, 000 7.5% gel is used. Molecular weight of protein Mr can be determined by the comparing its mobility with those of a number of protein used as standard. By plotting a graph of distance moved against log Mr for each of standard proteins, a calibration curve can be constructed. The distance moved by the protein of unknown Mr is then measured and its log Mr and hence Mr can be determined from calibration curve. A protein generally have single band in the protein unless it has two same subunit. d. Native (buffer) gel In this method SDS is not used for the separation of the protein subunit prior to loading. Here protein is separated according to the native charge of protein at pH of the gel (normally pH 8.7) and according to the different electrophoretic nobilities and the sieving effect of the gel. The method is generally used for the enzyme substrate reaction and total protein content. e. Gradient gel The protein is separated according to the gradient formed by the different concentration of the gel. Generally 5% at the top, while 15% gel is used at the bottom. The main advantage is that very similar molecular weight can be resolved. f. Iso-electric focusing: IEF gel: It utilizes the horizontal gels and separates the protein according to the isoelectric point of the protein. As we know that protein is the ampholytes, it can be separated easily. Here riboflavin is used for initiation of the polymerization of the gel. Protein having pH lower than iso-electric point will be positively charged, and will initially migrate towards the cathode. As they proceed further, the charge on the protein decreases slowly and at one point protein stops where IP is equal to the charge. For determining the IP of the protein, a standard IP of known protein can be determined and with the help of this calibration curve, IP of unknown protein can be determined. IEF is highly sensitive technique and used for studying heterogeneity of the protein. g. Chromatofocussing Is suitable for the protein separation. This is based on forming a pH gradient. The ion-exchanger is fixed in the column and set to a particular temperature and pH. The difference of pH lies between 3-4 unit from upper and bottom. On adding protein at top of column the protein moves at its isoelectric point. Just beyond the isoelectric point the protein is bind to the positive charge of the ionexchange column. Chromatofocussing gives a good resolution of quit complex mixture of protein provided that there is close difference in their isoelectric point, it gives poor resolution of compound having same isoelectric point.
2D-PAGE This technique utilizes the technique of both IEF and the SDS-PAGE. In the first dimension isoelectric focusing is done in polyacrylamide gel in narrow tubes in the presence of ampholytes, 8M urea and non-ionic detergent. Denatured protein is separated in the gel according to the isoelectric point. In another step the protein is run in presence of SDS. Generally the technique is used in the translational product of the gene or mRNA. It also helps in isolating the extra protein in the expression. Now a days to reduce so much of the labor, computerized 2D is done. DETECTION, ESTIMATION AND RECOVERY OF PROTEIN GELS. CBB or Coomassie blue R -250 is most commonly used for the detection of the protein in the gel. Staining is done in the 0.1% (w/v) CBB in methanol: water: glacial acetic acid. This acid methanol mixture acts as a denaturant to precipitate or fix the protein in the gel. This prevents the protein from being washed out while it is stained. Coomassie blue is highly sensitive, means it can detects the 0.1 g of protein. Silver stain is more sensitive. Ag+ ion is reduced to metallic silver on the protein, where the silver is deposited to give black band. It can be used immediately after the electrophoresis. It is 100 time more sensitive than CBB. Glycoprotein can be detected by the stain called as PAS (periodic acid stiff stain). It gives pink red bands. It is difficult to observe in the gel. Quantitative analysis of the protein can be done by the method called as scanning densitometry. Protein can be further analyzed and protein can be purified. Protein band can be cut out of protein and sequence by the gas phase sequencer. Note: separation of protein is called as western blotting, while separation of protein is called as southern blotting. i. AGAROSE GEL ELECTROPHORESIS OF DNA As we know that DNA is larger than proteins and therefore they are unable to enter the polyacrylamide gel. So the convenient way is to use agarose to analyze the DNA. Note that single stranded DNA is always expressed in nt (nucleotide) while double stranded DNA is expressed always in the base pair or kilo base pair. Agarose gel of 0.3% will separated double stranded DNA molecules of about 5 and 60 kb size, whereas 2% gels can be used for the separation of the sample between 0.1 and 3 kb. Most of the lab uses 0.8% gels which are suitable for separation for the DNA molecule in the range of 0.5 kb ---10 kb. The molecular weight of the DNA can be known from the calibration curve prepared from the known standard of the DNA. The smaller fragment of the DNA moves faster in the gel in compare to the larger fragments. Also open DNA moves slower in compare to the closed circular DNA. Preparation of the gel Gel is prepared by dissolving agarose in the water according to the need. The volume is calculated by measuring the length and breadth of the gel tank. The gel is boiled and poured in the tank and left for the solidifying. In the mean time, the comb is introduced in the gel, so that well can form in the gel. In this well, DNA material is loaded. The gel is sealed surrounding the gel tank which is later on opened before placing them in the electrophoresis tank. Electrophoresis tank is filled with the buffer. Along with the sample some dye and glycerol is also loaded so that they cant float in the buffer. Bromophenol blue is generally used and also ethidium bromide. Ethidium bromide is introduced in between the DNA double stranded DNA. Polyacrylamide gel Polymerization of the acrylamide results in the formation of the polyacrylamide. So they are also called as PAGE. This requires the presence of smaller amount of the N, N methylene bis acrylamide (bis-acrylamide). Actually the bis acrylamide is two acrylamide joined by the methylene. Polyacrylamide polymerization is the free radical catalysis, and is initiated by the addition of ammonium persulphate and the base N,N,N,N- TETRAMETHYLENE DIAMINE (TEMED). TEMED catalyses the decomposition of the persulphate ion to give a free radical (a molecule with
unpaired electron). It must be noted that all oxygen should be removed prior to use of the gel by degassing it in the vacuum. Induction of the polymerization can be done by a method called as photo-polymerization, but it needs riboflavin which generated the free radical. Acrylamide gel can be made with a content between 3 to 30 %. In SDS gel, 10% to 20 % acrylamide are used in techniques such as SDS gel electrophoresis.
Chapter -13 Enzyme Immobilization Introduction The word "immobilized enzyme" was coined by Katchalski-Katzir in 1971 (KatchalskiKatzir, 1993). Enzymes may be immobilized on solid carriers by various techniques, such as carrier-binding, cross-linking, or entrapment (Katchalski-Katzir, 1993). Immobilization requires carriers to bind and to support the enzymes. A variety of carriers have been tested with different enzymes. Some of these include alginate, magnetite, kappa-carrageenan, polyurethane, etc. There is sufficient variety to accommodate almost all available industrial enzymes. Interestingly, there are very few detailed studies to compare the efficacy of various immobilization methods or immobilization supports. It is generally believed that the best support for one enzyme may not work as well for another enzyme, and that each enzyme ought to be evaluated to determine the optimal carrier matrix system and conditions. Generally, the decision will depend on (a) the various characteristics of the enzymes; (b) requirements of the specific application, ie. the operational conditions; and (c) the properties and limitations of the support system (Bickerstaff, 1997). Aim of Enzyme Immobilization Enzyme immobilization is aimed to restrict the freedom of movement of an enzyme. When one wants to do enzyme immobilization, one must select the carrier (support, matrix) and decide on the method of immobilization. Carriers or support may be selected on the basis of the following considerations: [a] Physical properties Strength, available surface area, shape or form (eg. Beads, sheets, fibers), compressibility of carriers, porosity, pore volume, permeability, density, flow rate, pressure drop are the main requirements of the immobilization. [b] Chemical properties chemical property like hydrophilicity, inertness to enzyme(s), substrate(s) or cofactor(s), available functional groups for modification, ability to be regenerated or reused. Compatibility with certain buffers e.g. Alginate is not compatible with phosphate buffer. [c] Stability Immobilized enzyme must have stability on storage, residual enzyme activity on storage, mechanical stability of support material when subject to pressure or water flow. [d] Resistance Immobilized enzyme must be resistance against bacterial or fungal attack, disruption by chemicals, pH, temperature, organic solvents, and enzymes such as proteases. [e] Safety Immobilized enzyme must be safe from toxicity of component reagents, health and safety for process workers and end product users. Carriers must be "safe" if the end product is to be used for food, medical or pharmaceutical applications. For example, acrylamide is toxic. [f] Economic Availability and cost of carrier materials, chemicals, special equipment, reagents and technical skills is required. Beside this industrial scale chemical
preparation, feasibility for scale-up, continuous processing, effective working life, and re-usability must not be very costly. Reaction: Immobilized enzymes must not show any diffusion limitations on mass transfer of cofactors, substrates or products, Side reactions. Immobilized enzymes must not be biodegradability for example in waste treatment polyurethane is not naturally occurring and is not easily degraded. Therefore, its use may be undesirable for cleanup. Limitations of Immobilized Enzyme 1. An important factor determining the use of enzymes in a technological process is their expense. Several hundred enzymes are commercially available that are very costly, although some are much cheaper and many are much more expensive. As enzymes are catalytic molecules, they are not directly used up by the processes in which they are used. Their high initial cost, therefore, should only be incidental to their use. 2. However due to denaturation, they do lose activity with time. If possible, they should be stabilised against denaturation and utilised in an efficient manner. 3. When they are used in a soluble form, they retain some activity after the reaction which cannot be economically recovered for re-use and is generally wasted. This activity residue remains to contaminate the product and its removal may involve extra purification costs. In order to eliminate this wastage, and give an improved productivity, simple and economic methods must be used which enable the separation of the enzyme from the reaction product. The easiest way of achieving this is by separating the enzyme and product during the reaction using a two-phase system; one phase containing the enzyme and the other phase containing the product. The enzyme is imprisoned within its phase allowing its re-use or continuous use but preventing it from contaminating the product; other molecules, including the reactants, are able to move freely between the two phases. This is known as immobilisation and may be achieved by fixing the enzyme to, or within, some other material. The term 'immobilisation' does not necessarily mean that the enzyme cannot move freely within its particular phase, although this is often the case. A wide variety of insoluble materials, also known as substrates (not to be confused with the enzymes' reactants), may be used to immobilise the enzymes by making them insoluble. These are usually inert polymeric or inorganic matrices. Immobilisation of enzymes often incurs an additional expense and is only undertaken if there is a sound economic or process advantage in the use of the immobilised, rather than free (soluble), enzymes. Advantages of Immobilizations 1. The most important benefit derived from immobilisation is the easy separation of the enzyme from the products of the catalysed reaction. This prevents the enzyme contaminating the product, minimising downstream processing costs and possible effluent handling problems, particularly if the enzyme is noticeably toxic or antigenic. 2. It also allows continuous processes to be practicable, with a considerable saving in enzyme, labour and overhead costs. Immobilisation often affects the stability and activity of the enzyme, but conditions are usually available where these properties are little changed or even enhanced. 3. The productivity of an enzyme, so immobilised, is greatly increased as it may be more fully used at higher substrate concentrations for longer periods than the free enzyme. Insoluble immobilised enzymes are of little use, however, where any of the reactants are also insoluble, due to steric difficulties. Methods of immobilizations There are four principal methods available for immobilising enzymes (Figure 3.1): I. adsorption II. covalent binding III. entrapment IV. membrane confinement Figure 6.1. Immobilised enzyme systems. (a) enzyme non-covalently adsorbed to an
insoluble particle; (b) enzyme covalently attached to an insoluble particle; (c) enzyme entrapped within an insoluble particle by a cross-linked polymer; (d) enzyme confined within a semipermeable membrane. ________________________________________ Carrier matrices Carrier matrices for enzyme immobilisation by adsorption and covalent binding must be chosen with care. Of particular relevance to their use in industrial processes is their cost relative to the overall process costs; ideally they should be cheap enough to discard. The manufacture of high-valued products on a small scale may allow the use of relatively expensive supports and immobilisation techniques whereas these would not be economical in the large-scale production of low added-value materials. A substantial saving in costs occurs where the carrier may be regenerated after the useful lifetime of the immobilised enzyme. The surface density of binding sites together with the volumetric surface area sterically available to the enzyme, determine the maximum binding capacity. The actual capacity will be affected by the number of potential coupling sites in the enzyme molecules and the electrostatic charge distribution and surface polarity (i.e. the hydrophobic-hydrophilic balance) on both the enzyme and support. The nature of the support will also have a considerable affect on an enzyme's expressed activity and apparent kinetics. The form, shape, density, porosity, pore size distribution, operational stability and particle size distribution of the supporting matrix will influence the reactor configuration in which the immobilised biocatalyst may be used. The ideal support is cheap, inert, physically strong and stable. It will increase the enzyme specificity (kcat/Km) whilst reducing product inhibition, shift the pH optimum to the desired value for the process, and discourage microbial growth and non-specific adsorption. Some matrices possess other properties which are useful for particular purposes such as ferromagnetism (e.g. magnetic iron oxide, enabling transfer of the biocatalyst by means of magnetic fields), a catalytic surface (e.g. manganese dioxide, which catalytically removes the inactivating hydrogen peroxide produced by most oxidases), or a reductive surface environment (e.g. titania, for enzymes inactivated by oxidation). Clearly most supports possess only some of these features, but a thorough understanding of the properties of immobilised enzymes does allow suitable engineering of the system to approach these optimal qualities. Adsorption of enzymes In adsorption, the enzyme is bound to the carrier material via reversible surface interactions. The forces involved are electrostatic, such as van der Waals forces, ionic and H-bonding interaction, and possibly hydrophobic forces. The forces are generally weak, but there are sufficiently large to allow reasonable binding. The carriers with adsorption properties are selected on the basis of knowledge of their compatibility with the enzyme. Adsorption utilizes existing surface interactions between enzyme and carrier, and does not require chemical activation or modification. Normally, one does not see any damage to the enzyme by the carriers. Figure 6.2 showing the carrier that adsorbs the Enzyme Some advantages of adsorption include 1. Little or no damage to the enzyme. 2. Simple, cheap and immobilization can be done quickly. 3. No chemical changes to carriers or enzyme. 4. Easily reversible. Some disadvantages of adsorption include Desorption or leakage of enzyme from support. Desorption may occur on changing environmental conditions (such as pH, temperature, ionic strength) or on conformational changes arising from substrate/cofactor binding, contaminant binding, etc. Some physical factors such as flow rate, agitation by stirring,
collision or abrasion can cause desorption. Non-specific binding by substrate, cofactor or contaminants to the carrier may result in diffusion limitations and mass transfer problems. This may in turn change the kinetic properties of the reactions. Binding of protons to the support may change the pH of the microenvironment and change the reaction rate. Overloading of carrier possible. This may result in reduced catalytic activity. Possible steric hindrance by the carrier material. Adsorption of enzymes on to insoluble supports is a very simple method of wide applicability and capable of high enzyme loading (about one gram per gram of matrix). Simply mixing the enzyme with a suitable adsorbent, under appropriate conditions of pH and ionic strength, followed, after a sufficient incubation period, by washing off loosely bound and unbound enzyme will produce the immobilised enzyme in a directly usable form (Figure 3.2). The driving force causing this binding is usually due to a combination of hydrophobic effects and the formation of several salt links per enzyme molecule. The particular choice of adsorbent depends principally upon minimising leakage of the enzyme during use. Although the physical links between the enzyme molecules and the support are often very strong, they may be reduced by many factors including the introduction of the substrate. Care must be taken that the binding forces are not weakened during use by inappropriate changes in pH or ionic strength. Examples of suitable adsorbents are ion-exchange matrices (Table 3.1), porous carbon, clays, hydrous metal oxides, glasses and polymeric aromatic resins. Ion-exchange matrices, although more expensive than these other supports, may be used economically due to the ease with which they may be regenerated when their bound enzyme has come to the end of its active life; a process which may simply involve washing off the used enzyme with concentrated salt solutions and re-suspending the ion exchanger in a solution of active enzyme. ________________________________________ Figure 6.2. Schematic diagram showing the effect of soluble enzyme concentration on the activity of enzyme immobilised by adsorption to a suitable matrix. The amount adsorbed depends on the incubation time, pH, ionic strength, surface area, porosity, and the physical characteristics of both the enzyme and the support. ________________________________________ Table 6.1 Preparation of immobilised invertase by adsorption (Woodward 1985) Support type % bound at DEAE-Sephadex anion exchanger CM-Sephadex cation exchanger pH 2.5 0 100 pH 4.7 100 75 pH 7.0 100 34 ________________________________________ Covalent coupling This involves the formation of a covalent bond between the enzyme and the carrier material. The bond is normally formed between functional groups on the carrier and the enzyme. Those on the enzymes are usually amino acid residues such as amino (NH2) group from Lys or Arg, carboxyl (COOH) group from Asp, Glu, hydroxyl (OH) group from Ser, Thr, and sulfhydryl (SH) group from Cys. Tests must be run to ensure the formation of covalent bonds will not inactivate the enzyme. Through chemical modifications, functional groups on the carriers can be altered to different forms to accommodate different kinds of covalent bonds to be formed with the enzyme. For example, chemical modification of -OH group can give rise to AEcellulose (aminoethyl), CM- cellulose (carboxymethyl), and DEAE-cellulose (diethylaminoethyl). This increases the range of immobilization methods that can be used for a given carrier. While many supports exist, an important factor for enzyme immobilization appears to be hydrophilicity, which helps to maintain enzyme activity in a hydrophilic
milieu. Immobilisation of enzymes by their covalent coupling to insoluble matrices is an extensively researched technique. Only small amounts of enzymes may be immobilised by this method (about 0.02 gram per gram of matrix) although in exceptional cases as much as 0.3 gram per gram of matrix has been reported. The strength of binding is very strong, however, and very little leakage of enzyme from the support occurs. Functional groups that affects the covalent coupling The relative usefulness of various groups, found in enzymes, for covalent link formation depends upon their availability and reactivity (nucleophilicity), in addition to the stability of the covalent link, once formed (Table 3.2). The reactivity of the protein side-chain nucleophiles is determined by their state of protonation (i.e. charged status) and roughly follows the relationship -S- > -SH > -O- > -NH2 > -COO- > -OH >> -NH3+where the charges may be estimated from a knowledge of the pKa values of the ionising groups (Table 1.1) and the pH of the solution. Lysine residues are found to be the most generally useful groups for covalent bonding of enzymes to insoluble supports due to their widespread surface exposure and high reactivity, especially in slightly alkaline solutions. They also appear to be only very rarely involved in the active sites of enzymes. ________________________________________ Table 6.2 Relative usefulness of enzyme residues for covalent coupling Residue Content Exposure Reactivity Stability of couple Use Aspartate + ++ + + + Arginine + ++ Cysteine ++ Cystine + Glutamate + ++ + + + Histidine ++ + + + Lysine ++ ++ ++ ++ ++ Methionine Serine ++ + + Threonine ++ + Tryptophan Tyrosine + + _+ + C terminus ++ + + + N terminus ++ ++ ++ + Carbohydrate - ~ ++ ++ + + Others - ~ ++ - ~ ++ The most commonly used method for immobilising enzymes on the research scale (i.e. using less than a gram of enzyme) involves Sepharose, activated by cyanogen bromide. This is a simple, mild and often successful method of wide applicability. Sepharose is a commercially available beaded polymer which is highly hydrophilic and generally inert to microbiological attack. Chemically it is an agarose (poly{1,3-D-galactose--1,4-(3,6-anhydro)-L-galactose}) gel. The hydroxyl groups of this polysaccharide combine with cyanogen bromide to give the reactive cyclic imido-carbonate. This reacts with primary amino groups (i.e. mainly lysine residues) on the enzyme under mildly basic conditions (pH 9 - 11.5, Figure 3.3a). The high toxicity of cyanogen bromide has led to the commercial, if rather expensive, production of ready-activated Sepharose and the investigation of alternative methods, often involving chloroformates, to produce similar intermediates (Figure 3.3b). Carbodiimides (Figure 13.3c) are very useful bifunctional reagents as they allow the coupling of amines to carboxylic acids. Careful control of the reaction conditions and choice of carbodiimide allow a great degree of selectivity in this reaction. Glutaraldehyde is another bifunctional reagent which may be used to cross-link enzymes or link them to supports (Figure 13.3d). It is particularly useful for
producing immobilised enzyme membranes, for use in biosensors, by cross-linking the enzyme plus a non-catalytic diluent protein within a porous sheet (e.g. lens tissue paper or nylon net fabric). The use of trialkoxysilanes allows even such apparently inert materials as glass to be coupled to enzymes (Figure 13.3e). There are numerous other methods available for the covalent attachment of enzymes (e.g. the attachment of tyrosine groups through diazo-linkages, and lysine groups through amide formation with acyl chlorides or anhydrides). ________________________________________ A. cyanogen bromide [6.3] . (a) Activation of Sepharose by cyanogen bromide. Conditions are chosen to minimise the formation of the inert carbamate. (b) Ethyl chloroformate [6.3] (b) Chloroformates may be used to produce similar intermediates to those produced by cyanogen bromide but without its inherent toxicity. (c) Carbodiimide [6.3] (c) Carbodiimides may be used to attach amino groups on the enzyme to carboxylate groups on the support or carboxylate groups on the enzyme to amino groups on the support. Conditions are chosen to minimise the formation of the inert substituted urea. (d) Glutaraldehyde [6.3] (d) Glutaraldehyde is used to cross-link enzymes or link them to supports. It usually consists of an equilibrium mixture of monomer and oligomers. The product of the condensation of enzyme and glutaraldehyde may be stabilised against dissociation by reduction with sodium borohydride. (e) 3-aminopropyltriethoxysilane Figure 6.3. (e) The use of trialkoxysilane to derivatise glass. The reactive glass may be linked to enzymes by a number of methods including the use thiophosgene, as shown. ________________________________________ It is clearly important that the immobilised enzyme retains as much catalytic activity as possible after reaction. This can, in part, be ensured by reducing the amount of enzyme bound in non-catalytic conformations (Figure 3.4). Immobilisation of the enzyme in the presence of saturating concentrations of substrate, product or a competitive inhibitor ensures that the active site remains unreacted during the covalent coupling and reduces the occurrence of binding in unproductive conformations. The activity of the immobilised enzyme is then simply restored by washing the immobilised enzyme to remove these molecules. ________________________________________ Figure 6.4. The effect of covalent coupling, on the expressed activity of an immobilised enzyme,. (a) Immobilised enzyme, (E) with its active site unchanged and ready to accept the substrate molecule (S), as shown in (b). (c) Enzyme bound in a non-productive mode due to the inaccessibility of the active site. (d) Distortion of the active site produces an inactive immobilised enzyme. Nonproductive modes are best prevented by the use of large molecules reversibly bound in or near the active site. Distortion can be prevented by use of molecules which can sit in the active site during the coupling process, or by the use of a freely reversible method for the coupling which encourages binding to the most energetically stable (i.e. native) form of the enzyme. Both (c) and (d) may be reduced by use of 'spacer' groups between the enzyme and support, effectively displacing the enzyme away from the steric influence of the surface. ________________________________________ Entrapment and Encapsulation In both of these methods, the enzyme remains free in solution, but restricted in
movement by the lattice structure of a gel. The porosity of the gel lattice is controlled to ensure that the structure is tight enough to prevent leakage of enzyme, while allowing free movement of substrates, cofactors and products. Encapsulation generally refers to larger capsules that allow for co-immobilization of different combinations of enzymes for selected applications. Notably, red blood cells can be used as encapsulation capsules. Entrapment of enzymes Entrapment of enzymes within gels or fibers is a convenient method for use in processes involving low molecular weight substrates and products. Amounts in excess of 1 g of enzyme per gram of gel or fiber may be entrapped. However, the difficulty which large molecules have in approaching the catalytic sites of entrapped enzymes precludes the use of entrapped enzymes with high molecular weight substrates. The entrapment process may be a purely physical caging or involve covalent binding. As an example of this latter method, the enzymes' surface lysine residues may be derivitized by reaction with acryloyl chloride (CH2=CH-CO-Cl) to give the acryloyl amides. This product may then be copolymerised and cross-linked with acrylamide (CH2=CH-CO-NH2) and bisacrylamide (H2N-CO-CH=CHCH=CH-CO-NH2) to form a gel. Enzymes may be entrapped in cellulose acetate fibers by, for example, making up an emulsion of the enzyme plus cellulose acetate in methylene chloride, followed by extrusion through a spinneret into a solution of an aqueous precipitant. Entrapment is the method of choice for the immobilisation of microbial, animal and plant cells, where calcium alginate is widely used. Membrane confinement Membrane confinement of enzymes may be achieved by a number of quite different methods, all of which depend for their utility on the semipermeable nature of the membrane. This must confine the enzyme whilst allowing free passage for the reaction products and, in most configurations, the substrates. The simplest of these methods is achieved by placing the enzyme on one side of the semipermeable membrane whilst the reactant and product stream is present on the other side. Hollow fiber membrane units are available commercially with large surface areas relative to their contained volumes (> 20 m2 l-1) and permeable only to substances of molecular weight substantially less than the enzymes. Although costly, these are very easy to use for a wide variety of enzymes (including regenerating coenzyme systems, see Chapter 8) without the additional research and development costs associated with other immobilisation methods. Encapsulation of Enzymes Enzymes, encapsulated within small membrane-bound droplets or liposomes, may also be used within such reactors. As an example of the former, the enzyme is dissolved in an aqueous solution of 1,6-diaminohexane. This is then dispersed in a solution of hexanedioic acid in the immiscible solvent, chloroform. The resultant reaction forms a thin polymeric (Nylon-6,6) shell around the aqueous droplets which traps the enzyme. Liposomes are concentric spheres of lipid membranes, surrounding the soluble enzyme. They are formed by the addition of phospholipids to enzyme solutions. The micro-capsules and liposomes are washed free of non-confined enzyme and transferred back to aqueous solution before use. Table 6.3 presents a comparison of the more important general characteristics of these methods. ________________________________________ Table 6.3 Generalised comparison of different enzyme immobilisation techniques. Characteristics Adsorption Covalent binding Entrapment Membrane confinement Preparation Simple Difficult Difficult Simple Cost Low High Moderate High Binding force Variable Strong Weak Strong Enzyme leakage Yes No Yes No Applicability Wide Selective Wide Very wide Running Problems High Low High High Matrix effects Yes Yes Yes No
Large diffusional barriers No No Yes Yes Microbial protection No No Yes Yes ________________________________________ ________________________________________ There are several methods of immobilization (Bickerstaff, 1997), as listed below. Methods compatible with the enzyme, substrate or cofactor should be selected. For example, the entrapment method may not work well with cellulose substrate because the substrate itself is large and will not readily go into the entrapment matrix to reach the enzyme. Crosslinking This method joins the enzyme to each other to form a large, 3-D structure without any support. This can be done through chemical (covalent bonding) or physical means (flocculation). Tests must be done to ensure the active site remains free and available for catalytic activity. Encapsulation often improves the operational stability and sometimes efficiency of enzyme catalysis under industrial conditions, and also allows their easy separation from the medium. This allows for easier recovery of the enzymes for repeat use. The improved stability and ease of separation of immobilized enzymes make them suitable to be used in a variety of bioreactors. They are also easier to scale up. In addition, encapsulation allows for ease of handling and storage of enzymes. Sometimes other properties of the enzyme may be altered by immobilization, e.g. altered pH-activity profile.
Chapter-14 Introduction to Industrial biotechnology Industrial biotechnology applies the techniques of modern molecular biology to improve the efficiency and reduce the environmental impacts of processes in industries like food production, grain cleaning, textiles, paper and pulp and specialty chemicals. Just as biotechnology is transforming the pharmaceutical industry, some observers predict the same impact in the industrial sector. Today up to 90% of the enzymes used in large scale, for commercial applications result from the exploitation of rDNA methods in the manufacturing process or for the improvement of the catalysts themselves. The progress made in applying the techniques of genetic engineering in the development of industrial-scale processes that produce or utilize enzymes has been simply amazing. Both improved economics as well as the manufacture of novel products not possible or practical by traditional chemical approaches have been achieved. Even today, much of the science is focused on the development of new technological approaches that will allow the future solution of problems of fundamental understanding as well as practical application. Industrial biotechnology companies develop biocatalysts, such as enzymes, to be used in chemical synthesis. Enzymes are proteins produced by all living organisms. In humans, enzymes help digest food, signal cells to turn on and off and perform other complex functions. Other animals use enzymes to break cellulose into sugar or break up proteins. Scientists can locate enzymes in the natural environment with special functional characteristics that have significant commercial value. Using biotechnology, the desired enzyme can then be manufactured in commercial quantities. Most enzymes are manufactured in fermentation systems like human therapeutic proteins. The manufacturing process uses renewable resources as a raw material feedstock. Contrary to inorganic catalysts such as acids, bases, metals and metal oxides, enzymes are very specific. In other words, each enzyme can break down or synthesize one particular compound. In some cases, they limit their action to specific bonds in the compounds with which they react. Most proteases, for instance, can break down several types of protein, but in each protein molecule
only certain bonds will be cleaved depending on which enzyme is used. In industrial processes, the specific action of enzymes allows high yields to be obtained with a minimum of unwanted byproducts. Enzymes are very efficient catalysts. For example, the enzyme catalase, which is found abundantly in the liver and red blood cells, is so efficient that in one minute one enzyme molecule can catalyze the breakdown of five million molecules of hydrogen peroxide into water and oxygen.Being formed to work in living cells, enzymes can work at atmospheric pressure and in mild conditions in terms of temperature and acidity. Most enzymes function optimally at a temperature of 30-70C and at pH values which are near pH 7. For certain technical applications, special enzymes have been developed that work at higher temperatures. However, no enzyme can withstand temperatures above 100C for long. Enzyme processes are therefore potentially energysaving and save investing in special equipment resistant to heat, pressure or corrosion. Due to their efficiency, specific action, the mild conditions in which they work and their high biodegradability, enzymes are very well suited to a wide range of industrial applications.Nature provides rich resources for innovative solutions for many industries. Companies involved in industrial biotechnology are constantly striving to discover and develop high-value enzymes and bioactive compounds that will enhance current industrial processes. Chemical processes, including paper manufacturing, textile processing, oil field chemicals and specialty chemical synthesis reactions, sometimes need to run at very high or very low temperatures or high or low pHs. The challenge is to find organisms that can survive and even thrive in these environments. The opportunity to identify unique bioactive molecules is vast. Less than 1 percent of the microorganisms in the world have been cultured and characterized. Application of Immobilised-Enzyme Processes Introduction Immobilise -enzyme systems are used where they offer cost advantages to users on the basis of total manufacturing costs. The plant size needed for continuous processes is two orders of magnitude smaller than that required for batch processes using free enzymes. The capital costs are, therefore, considerably smaller and the plant may be prefabricated cheaply off-site Immobilised enzymes offer greatly increased productivity on an enzyme weight basis and also often provide process advantages (see Chapter 6) Currently used immobilised-enzyme processes are given in Table 7.1. ________________________________________ Table 7.1 Some of the more important industrial uses of immobilised enzymes Enzyme EC number Product Aminoacylase 3.5.1.14 L-Amino acids Aspartate ammonia-lyase 4.3.1.1 L-Aspartic acid Aspartate 4-decarboxylase 4.1.1.12 L-Alanine Cyanidase 3.5.5.x Formic acid (from waste cyanide) Glucoamylase 3.2.1.3 D-Glucose Glucose isomerase 5.3.1.5 High -fructose corn syrup Histidine ammonia-lyase 4.3.1.3 Urocanic acid Hydantoinasea 3.5.2.2 D- and L-amino acids Invertase 3.2.1.26 Invert sugar Lactase 3.2.1.23 Lactose-free milk and whey Lipase 3.1.1.3 Cocoa butter substitutes Nitrile hydratase 4.2.I.x Acrylamide Penicillin amidases 3.5.1.11 Penicillins Raffinase 3.2.1.22 Raffinose-free solutions Thermolysin 3.2.24.4 Aspartame a Dihydropyrimidinase 1-High-fructose corn syrups (HFCS) With the development of glucoamylase in the 1940s and 1950s it became a
straightforward matter to produce high DE glucose syrups. However, these have shortcomings as objects of commerce: D-glucose has only about 70% of the sweetness of sucrose, on a weight basis, and is comparatively insoluble. Batches of 97 DE glucose syrup at the final commercial concentration (71% (w/w)) must be kept warm to prevent crystallisation or diluted to concentrations that are microbiologically insecure. Fructose is 30% sweeter than sucrose, on a weight basis, and twice as soluble as glucose at low temperatures so a 50% conversion of glucose to fructose overcomes both problems giving a stable syrup that is as sweet as a sucrose solution of the same concentration (see Table 14.3). The isomerisation is possible by chemical means but not economical, giving tiny yields and many by-products (e.g. 0.1 M glucose 'isomerised' with 1.22 M KOH at 5C under nitrogen for 3.5 months gives a 5% yield of fructose but only 7% of the glucose remains unchanged, the majority being converted to various hydroxy acids). 2-GLUCOSE ISOMERASE Glucose is normally isomerised to fructose during glycolysis but both sugars are phosphorylated. The use of this phosphohexose isomerase may be ruled out as a commercial enzyme because of the cost of the ATP needed to activate the glucose and because two other enzymes (hexokinase and fructose-6-phosphatase) would be needed to complete the conversion. Only an isomerase that would use underivatised glucose as its substrate would be commercially useful but, until the late 1950s, the existence of such an enzyme was not suspected. At about this time, enzymes were found that catalyse the conversion of D-xylose to an equilibrium mixture of D-xylulose and D-xylose in bacteria. When supplied with cobalt ions, these xylose isomerases were found to isomerise -D-glucopyranose to -D fructofuranose. Now it is known that several genera of microbes, mainly bacteria, can produce such glucose isomerases: The commercial enzymes are produced by Actinoplanes missouriensis, Bacillus coagulans and various Streptomyces species; as they have specificities for glucose and fructose which are not much different from that for xylose and ways are being found to avoid the necessity of xylose as inducer, these should perhaps now no longer be considered as xylose isomerases. They are remarkably friendly enzymes in that they are resistant to thermal denaturation and will act at very high substrate concentrations, which have the additional benefit of substantially stabilising the enzymes at higher operational temperatures. The vast majority of glucose isomerases are retained within the cells that produce them but need not be separated and purified before use. All glucose isomerases are used in immobilised forms. Although differerent immobilisation methods have been used for enzymes from differerent organisms, the principles of use are very similar. Immobilisation is generally by cross-linking with glutaraldehyde, plus in some cases a protein diluent, after cell lysis or homogenisation. Originally, immobilised glucose isomerase was used in a batch process. This proved to be costly as the relative reactivity of fructose during the long residence times gave rise to significant by -product production. Also, difficulties were encountered in the removal of the added Mg2+ and Co2+ and the recovery of the catalyst. Nowadays most isomerisation is performed in PBRs (Table 14.2). They are used with high substrate concentration (35-45% dry solids, 93-97% glucose) at 5560C. The pH is adjusted to 7.5-8.0 using sodium carbonate and magnesium sulphate is added to maintain enzyme activity (Mg2+ and Co2+ are cofactors). The Ca2+ concentration of the glucose feedstock is usually about 25 m, left from previous processing, and this presents a problem. Ca2+ competes successfully for the Mg2+ binding site on the enzyme, causing inhibition. At this level the substrate stream is normally made 3 mM with respect to Mg2+. At higher concentrations of calcium a Mg2+ : Ca2+ ratio of 12 is recommended. Excess Mg2+ is uneconomic as it adds to the purification as well as the isomerisation costs. The need for Co2+ has not been eliminated altogether, but the immobilisation methods now used fix the cobalt ions so that none needs to be added to the substrate streams. ________________________________________
Table 7.2. Comparison of glucose isomerisation methods Parameter Batch (soluble GI) Batch (immobilised Gl) Continuous (PBR) Reactor volume (m3) 1100 1100 15 Enzyme consumption (tonnes) 180 11 2 Activity, half-life (h) 30 300 1500 Active life, half-lives 0.7 2 3 Residence time (h) 20 20 0.5 Co2+ (tonnes) 2 1 0 Mg2+ (tonnes) 40 40 7 Temperature (C) 65 65 60 pH 6.8 6.8 7.6 Colour formation (A420) 0.7 0.2 < 0.1 Product refining Filtration C-treatmenta Cation exchange Anion exchange C-treatment Cation exchange Anion exchange C -treatment Capital, labour and energy costs, tonne-1 5 5 1 Conversion cost, tonne-1 500 30 5 All processes start with 45% (w/w) glucose syrup DE 97 and produce 10000 tonnes per month of 42% fructose dry syrup. Some of the improvement that may be seen for PBR productivity is due to the substantial development of this process. a Treatment with activated carbon. ________________________________________ Precautions It is essential for efficient use of immobilised glucose isomerase that the substrate solution is adequately purified so that it is free of insoluble material and other impurities that might inactivate the enzyme by chemical (inhibitory) or physical (pore-blocking) means. In effect, this means that glucose produced by acid hydrolysis cannot be used, as its low quality necessitates extensive and costly purification. Insoluble material is removed by filtration, sometimes after treatment with flocculants, and soluble materials are removed by ion exchange resins and activated carbon beads. This done, there still remains the possibility of inhibition due to oxidised by-products caused by molecular oxygen. This may be removed by vacuum de-aeration of the substrate at the isomerisation temperature or by the addition of low concentrations (< 50 ppm) of sulphite. At equilibrium at 60C about 51 % of the glucose in the reaction mixture is converted to fructose. However, because of the excessive time taken for equilibrium to be attained and the presence of oligosaccharides in the substrate stream, most manufacturers adjust flow rates so as to produce 42-46% (w/w) fructose (leaving 47-51 % (w/w) glucose). To produce 100 tonnes (dry substance) of 42% HFCS per day, an enzyme bed volume of about 4 m3 is needed. Activity decreases, following a first-order decay equation. The half-life of most enzyme preparations is between 50 and 100 days at 55C. Typically a batch of enzyme is discarded when the activity has fallen to an eighth of the initial value (i.e. after three half -lives). To maintain a constant fructose content in the product, the feed flow rate is adjusted according to the enzyme activity. Several reactors containing enzyme preparations of different ages are needed to maintain overall uniform production by the plant (Figure 14.10). In its lifetime 1 kg of
immobilised glucose isomerase (exemplified by Novo's Sweetzyme T) will produce 10 -11 tonnes of 42% fructose syrup (dry substance). ________________________________________ Figure 7.10. Diagram showing the production rate of a seven-column PBR facility on start -up, assuming exponential decay of reactor activity. The columns are brought into use one at a time. At any time a maximum of six PBRs are operating in parallel, whilst the seventh, exhausted, reactor is being refilled with fresh biocatalyst. PBR activities allowed to decay through three half-lives (to 12.5% initial activity) before replacement. The final average productivity is 2.51 times the initial productivity of one column. - - -- - - - PBR activities allowed to decay through two half-lives (to 25% initial activity) before replacement. The final average productivity is 3.23 times the initial productivity of one column. It may be seen that the final average production rate is higher when the PBRs are individually operated for shorter periods but this 29% increase in productivity is achieved at a cost of 50% more enzyme, due to the more rapid replacement of the biocatalyst in the PBRs. A shorter PBR operating time also results in a briefer start-up period and a more uniform productivity. ________________________________________ After isomerisation, the pH of the syrup is lowered to 4 - 5 and it is purified by ion-exchange chromatography and treatment with activated carbon. Then, it is normally concentrated by evaporation to about 70% dry solids.For many purposes a 42% fructose syrup is perfectly satisfactory for use but it does not match the exacting criteria of the quality soft drink manufacturers as a replacement for sucrose in acidic soft drinks. For use in the better colas, 55% fructose is required. This is produced by using vast chromatographic columns of zeolites or the calcium salts of cation exchange resins to adsorb and separate the fructose from the other components. The fractionation process, although basically very simple, is only economic if run continuously. The fructose stream (90% (w/w) fructose, 9% glucose) is blended with 42% fructose syrups to give the 55% fructose (42% glucose) product required. The glucose-rich 'raffinate' stream may be recycled but if this is done undesirable oligosaccharides build up in the system. Immobilised glucoamylase is used in some plants to hydrolyse oligosaccharides in the raffinate; here the substrate concentration is comparatively low (around 20% dry solids) so the formation of isomaltose by the enzyme is insignificant.Clearly the need for a second large fructose enrichment plant in addition to the glucose isomerase plant is undesirable and attention is being paid to means of producing 55% fructose syrups using only the enzyme. The thermodynamics of the system favour fructose production at higher temperatures and 55% fructose syrups could be produced directly if the enzyme reactors were operated at around 95C. The use of miscible organic co -solvents may also produce the desired effect. Both these alternatives present a more than considerable challenge to enzyme technology! The present world market for HFCS is over 5 million tonnes of which about 60% is for 55% fructose syrup with most of the remainder for 42% fructose syrup. This market is still expanding and ensures that HFCS production is the major application for immobilised-enzyme technology.The high-fructose syrups can be used to replace sucrose where sucrose is used in solution but they are inadequate to replace crystalline sucrose. Another ambition of the corn syrup industry is to produce sucrose from starch. This can be done using a combination of the enzymes phosphorylase (EC 2.4.1.1), glucose isomerase and sucrose phosphorylase (EC 2.4.1.7), but the thermodynamics do not favour the conversion so means must be found of removing sucrose from the system as soon as it is formed. This will not be easy but is achievable if the commercial pull (i.e. money available) is sufficient: phosphorylase starch (Gn) + orthophosphate glucose isomerase starch (Gn-1) + -glucose-1-phosphate [7.1]
glucose fructose [7.2] sucrose phosphorylase -glucose-1 -phosphate + fructose sucrose + orthophosphate [7.3] A further possible approach to producing sucrose from glucose is to supply glucose at high concentrations to microbes whose response to osmotic stress is to accumulate sucrose intracellularly. Provided they are able to release sucrose without hydrolysis when the stress is released, such microbes may be the basis of totally novel processes. 3-Use of immobilised raffinase The development of a raffinase (-D-galactosidase) suitable for commercial use is another triumph of enzyme technology. Plainly, it would be totally unacceptable to use an enzyme preparation containing invertase to remove this material during sucrose production. It has been necessary to find an organism capable of producing an -galactosidase but not an invertase. A mould, Mortierella vinacea var. raffinoseutilizer, fills the requirements. This is grown in a particulate form and the particles harvested, dried and used directly as the immobilised-enzyme preparation. It is stirred with the sugar beet juice in batch stirred tank reactors. When the removal of raffinose is complete, stirring is stopped and the juice pumped off the settled bed of enzyme. Enzyme, lost by physical attrition, is replaced by new enzyme added with the next batch of juice. The galactose released is destroyed in the alkaline conditions of the first stages of juice purification and does not cause any further problems while the sucrose is recovered. This process results in a 3% increase in productivity and a significant reduction in the costs of the disposal of waste molasses. Immobilised raffinase may also be used to remove the raffinose and stachyose from soybean milk. These sugars are responsible for the flatulence that may be caused when soybean milk is used as a milk substitute in special diets. 4-Use of immobilised Invertase Invertase was probably the first enzyme to be used on a large scale in an immobilised form (by Tate & Lyle). In the period 1941 -1946 the acid, previously used in the manufacture of Golden Syrup, was unavailable, so yeast invertase was used instead. Yeast cells were autolysed and the autolysate clarified by adjustment to pH 4.7, followed by filtration through a bed of calcium sulphate and adsorption into bone char. A layer of the bone char containing invertase was included in the bed of bone char already used for decolourising the syrup. The scale used was large, the bed of invertase-char being 2 ft (60 cm) deep in a bed of char 20 ft (610 cm) deep. The preparation was very stable, the limiting factors being microbial contamination or loss of decolourising power rather than loss of enzymic activity. The process was cost-effective but, not surprisingly, the product did not have the subtlety of flavour of the acid-hydrolysed material and the immobilised enzyme process was abandoned when the acid became available once again. Recently, however, it has been relaunched using BrimacTM, where the invertase -char mix is stabilised by cross-linking and has a half-life of 90 days in use (pH 5.5, 50C). The revival is due, in part, to the success of HFCS as a high-quality low-colour sweetener. It is impossible to produce inverted syrups of equivalent quality by acid hydrolysis. Enzymic inversion avoids the high-colour, high salt-ash, relatively low conversion and batch variability problems of acid hydrolysis. Although free invertase may be used (with residence times of about a day), the use of immobilised enzymes in a PBR (with residence time of about 15 min) makes the process competitive; the cost of 95% inversion (at 50% (w/w)) being no more than the final evaporation costs (to 75% (w/w)). A productivity of 16 tonnes of inverted syrup (dry weight) may be achieved using one litre of the granular enzyme. 5-Production of amino acids Another early application of an immobilised enzyme was the use of the aminoacylase from Aspergillus oryzae to resolve racemic mixtures of amino acids. Figure [7.4] Chemically synthesised racemic N-acyl-DL-amino acids are hydrolysed
at pH 8.5 to give the free L-amino acids plus the unhydrolysed N-acyl-D-amino acids. These products are easily separated by differential crystallisation and the N-acyl-D-amino acids racemised chemically (or enzymically) and reprocessed. The enzyme is immobilised by adsorption to anion exchange resins (e.g. DEAE-Sephadex) and has an operational half-life of about 65 days at 50C in PBRs with residence times of about 30 min. The reactors may be re-activated in situ by simply adding more enzyme. The immobilised enzyme has proved a more economical process than the use of free enzyme mainly due to the more efficient use of the substrate and reductions in the cost of enzyme and labour. Novel and natural L-amino acids can be produced by the chemical conversion of aldehydes through DL-amino nitrites to racemic DL-hydantoins (reaction scheme [14.5]) followed by enzymic hydrolysis with hydantoinase and a carbamoylase (reaction scheme [14.6]) at pH 8.5. Both enzymes may be obtained from Arthrobacter species. D -Amino acids are important constituents in antibiotics and insecticides. They may be produced in a manner similar to the L-amino acids but using hydantoinases of differing specificity. The Pseudomonas striata enzyme is specific for Dhydantoins, allowing their specific hydrolysis to D-carbamoyl amino acids which can be converted to the D-amino acids by chemical treatment with nitrous acid. They remaining L-hydantoin may be simply racemised by base and the process repeated. [7.5] [7.6] L -Aspartic acid is widely used in the food and pharmaceutical industries and is needed for the production of the low -calorific sweetener aspartame. It may be produced from fumaric acid by the use of the aspartate ammonia-lyase (aspartase) from Escherichia coli. aspartate ammonia-lyase -OOCCH=CHCOO- + NH4+ -OOCCH2CH(NH3+)COO[7.7] fumaric acid L-aspartic acid A crude immobilised aspartate ammonia-lyase (50000 U g-1) may be prepared by entrapping Escherichia coli cells in a -carageenan gel crosslinked with glutaraldehyde and hexamethylenediamine. The process is operated in a PBR at pH 8.5 using ammonium fumarate as the substrate, with a reported operational halflife of 680 days at 37C. Urocanic acid is a sun-screening agent which may be produced from L-histidine by the histidine ammonia -lyase (histidase) from Achromobacter liquidum . The organism cannot be used directly as it has urocanate hydratase activity, which removes the urocanic acid. However, a brief heat treatment (70C, 30 min) inactivates this unwanted activity but has little effect on the histidine ammonialyase. A crude immobilised-enzyme preparation consisting of heat -treated cells entrapped in a polyacrylamide gel has been used to effect this conversion, showing a half-life of 180 days at 37C. 6- Use of immobilised lactase Lactase is one of relatively few enzymes that have been used both free and immobilised in large-scale processes. The reasons for its utility has been given earlier , but the relatively high cost of the enzyme is an added incentive for its use in an immobilised state.Immobilised lactases are important mainly in the treatment of whey, as the fats and proteins in the milk emulsion tend to coat the biocatalysts. This both reduces their apparent activity and increases the probability of microbial colonisation. Yeast lactase has been immobilised by incorporation into cellulose triacetate fibres during wet spinning, a process developed by Snamprogetti S.p.A. in Italy. The fibres are cut up and used in a batchwise STR process at 5C (Kluyveromyces lactis, pH optimum 6.4 -6.8, 90 U g-1). Fungal lactases have been immobilised on 0.5 mm diameter porous silica (35 nm mean pore diameter) using glutaraldehyde and -aminopropyltriethoxysilane (Asperigillus niger, pH optimum 3.0 -3.5, 500 U g-1; A. oryzae, pH optimum 4.0 -1.5, 400 U g-1). They are used in PBRs. Due to the
different pH optima of fungal and yeast lactases, the yeast enzymes are useful at the neutral pH of both milk and sweet whey, whereas fungal enzymes are more useful with acid whey. Immobilised lactases are particularly affected by two inherent short-comings. Product inhibition by galactose and unwanted oligosaccharide formation are both noticeable under the diffusion-controlled conditions usually prevalent. Both problems may be reduced by an increase in the effectiveness factor and a reduction in the degree of hydrolysis or initial lactose concentration, but such conditions also lead to a reduction in the economic return. The control of microbial contamination within the bioreactors is the most critical practical problem in these processes. To some extent, this may be overcome by the use of regular sanitation with basic detergent and a dilute protease solution. .7 Production of antibiotics Benzylpenicillins and phenoxymethylpenicillins (penicillins G and V, respectively) are produced by fermentation and are the basic precursors of a wide range of semisynthetic antibiotics, e.g. ampicillin. The amide link may be hydrolysed conventionally but the conditions necessary for its specific hydrolysis, whilst causing no hydrolysis of the intrinsically more labile but pharmacologically essential -lactam ring, are difficult to attain. Such specific hydrolysis may be simply achieved by use of penicillin amidases (also called penicillin acylases). Different enzyme preparations are generally used for the hydrolysis of the penicillins G and V, pencillin-V-amidase being much more specific than pencillinG-amidase. Penicillin amidase may be obtained from E. coli and has been immobilised on a number of supports including cyanogen bromide-activated Sephadex G200. It represents one of the earliest successful processes involving immobilised enzymes and is generally used in batch or semicontinuous STR processes (40,000 Ukg1penicillin G, 35C, pH 7.8, 2 h) where it may be reused over 100 times. It has also been used in PBRs, where it has an active life of over 100 days, producing about two tonnes of 6-aminopenicillanic acid kg-1of immobolised enzyme. [7.8] [7.9] The penicillin-G-amidases may be used 'in reverse' to synthesise penicillin and cephalosporin antibiotics by non -equilibrium kinetically controlled reactions. Ampicillin has been produced by the use of penicillin-G-amidase immobilised by adsorption to DEAE -cellulose in a packed bed column: [7.10] Many other potential and proven antibiotics have been synthesised in this manner, using a variety of synthetic -lactams and activated carboxylic acids. Preparation of acrylamide Acrylamide is an important monomer needed for the production of a range of economically useful polymeric materials. It may be produced by the addition of water to acrylonitrile. CH2=CHCN + H2O CH2=CHCONH2 [14.11] This process may be achieved by the use of a reduced copper catalyst (Cu+); however, the yield is poor, unwanted polymerisation or conversion to acrylic acid (CH2=CHCOOH) may occur at the relatively high temperatures involved (80 -140C) and the catalyst is difficult to regenerate. These problems may be overcome by the use of immobilised nitrile hydratase (often erroneously called a nitrilase). The enzyme from Rhodococcus has been used by the Nitto Chemical Industry Co. Ltd, as it contains only very low amidase activity which otherwise would produce unwanted acrylic acid from the acrylamide. Immobilised nitrile hydratase is simply prepared by entrapping the intact cells in a cross-linked 10% (w/v) polyacrylamide/dimethylaminoethylmethacrylate gel and granulating the product. It is used at 10C and pH 8.0-8.5 in a semibatchwise process, keeping the substrate acrylonitrile concentration below 3% (w/v). Using
1% (w/v) immobilised-enzyme concentration (about 50,000 U l-1) the process takes about a day. Product concentrations of up to 20% (w/v) acrylamide have been achieved, containing negligible substrate and less than 0.02% (w/w) acrylic acid. Acrylamide production using this method is about 4000 tonnes per year. The closely related enzymes cyanidase and cyanide hydratase are used to remove cyanide from industrial waste and in the detoxification of feeds and foodstuffs containing amygdalin (see equation [7.12]). HCN + 2H2O HCOO- + NH4+ [7.12] HCN + H2O HCONH2 [7.13]
Chapter-15 Application of Enzyme in clinical diagnosis and Industries Introduction Most important aspects of enzyme activities is the clinical diagnosis. Before 1940 ,only hydrolytic enzyme were used such as lipase, amylase, phosphatase, Trypsin, and pepsin and in diagnosis only 5% enzyme were in the use. Now a day up to 25% of the enzyme is used today for the clinical diagnosis. This is due to increased knowledge in the metabolic pathway and the role of enzyme. Due to enhancing in knowledge of enzyme it is possible to use it for rationale drug design to inhibit specific enzyme. The major application in the clinical field is the determination of the concentration of the substrate secreted in the body fluids with the help of enzymes and comparing it with the normal fluid concentration. With discoveries of monoclonal antibodies different specific detection become easier with the help of different assay system like ELISA. Development of medical applications for enzymes have been at least as extensive as those for industrial applications, reflecting the magnitude of the potential rewards: for example, pancreatic enzymes have been in use since the nineteenth century for the treatment of digestive disorders. The variety of enzymes and their potential therapeutic applications are considerable. A selection of those enzymes which have realised this potential to become important therapeutic agents is shown in Table 8.1 . At present, the most successful applications are extracellular: purely topical uses, the removal of toxic substances and the treatment of lifethreatening disorders within the blood circulation. Determination of enzyme activities For the determination of enzyme activities for clinical diagnosis, the most important thing is the availability of enzyme in the blood urine or in the tissue. Most of the clinical assay is done with the help of serum. Urine can also be used. Other body fluids that can be used is the body pleura, peritoneum, pericardium, cerebrospinal canal, synovia, stomoach, deudonium, semen , vagina, RBC and WBC. Activities of the enzyme in the serum may increases due to several reason. For example, due to tissue damage, due to increase cell turnover number, cellular proliferation, and neoplasia. In the disease state a tissue become inflamed and swelling occur and it become nacrotic. In that state it may secrete large amount of the enzymes from the dead cells for example release of glutamate dehydrogenase from the dead mitochondrial cells.Note that the stay of the release enzyme is very sort. For example glutamate S transferases has a half lie of 90 minute. To know amount of or fate of serum enzyme labeled DH injected in the rabbit. Then it has been observed that they were rapidly degraded and removed from the blood plasma. There are some tissue specific enzyme that may be detected from them. For example; acid phosphatases in the prostate and acetylcholineestarase in erythrocyte, .with the elctrophoresis technique we can determine many more enzyme
in the serum like alkaline phosphatase, amylase , creatine kinase, ceruloplasmine, , glucose-6 phosphate,dehydrogenase and aspartate aminotransferase. Other most studied enzyme in the blood serum is the LDH lactate dehydrogenase, creatine kinase, and alkaline phosphates. In forensic science main enzyme that is used is the adenosine deaminase, adenylate kinase, carbonate dehydrates and phosphatase esterase, phosphoglucomutase amino peptidase, and lactooylglutathione lyse. 1- Clinical Enzymology of liver disease. The most common disease of the liver is the hepatitis, cirrosis, tumors infection. The enzyme most used in the diagnosis is the (ST or SGOT) aspartate amino transferase, ALT or alanine aminotransferase or serum glutamate pyruvate transaminase(SPGT) , alkaline phosphatase, and - glutamyl transerase. Other enzyme is the lactate dehydrogenase, isocitrate dehydrogense 5 nucleotide and glutathione transferase. Ortnithin carbamoyltransferase is almost exclusive to liver. Serum alkaline phosphatase is mainly important in the liver disease. In the case of cirrhosis or hepatitis, aminotransferase are monitored for several months to follow progress of disease. Obstructive jaundice can be determined by the measurement of aminotransferases and alkaline phosphatase. Sometime these enzyme also secreted in the hepatitis so to decide which is the disease better to measure the ratio. The ratio of glutamate dehydrogenase / aminotranserase changes after one week of the viral hepatitis. The ratio is higher in the case of obstructive jaundice than in viral hepatitis. -glutamyltransferase used extensively in the liver specially in the case of alcoholism. 2- In Heart disease The main disease of the heart is the myocardial infarction and, nacrosis of the heart tissue. Enzymes are released during the nacrosis into the plasma. The three enzymes that is assayed are creatine kinase, aspartate aminotransferase, and lactate dehydrogenase. The first enzyme that was used in the diagnosis of the heart disease was the aspartate aminotransferase, but now creatine kinase, lactate dehydrogenase is used mostly. -amylase -amylase is an endoamylase and hydrolyze amylopectine. Its highest concentration is found in the intestine and salivary gland. In normal person its concentration is detected in low concentration in the urine and the serum but it is increases many fold during the pencreatitis. Pains occur severely in the upper duodenum. During the pancreatic carcinoma it is found in higher concentration in the pancreas. Also this enzyme is detected during the duodenal ulcers. amylase activity also increases during the mumps, when salivary glands become inflamed. Creatine Kinase and fructose bisphosphate aldolase. Creatine kinase predominantly occurs in the skeletal muscle, cardiac muscle and brain. Its concentration (creatine kinase) raised during brain stroke. Alkaline phosphates This enzyme is detected in the obstructive jaundice. It is also useful in the detection of bone disease like ostomalacia, rickets and hyperparathyroidism. This enzyme is normally found in the serum. This enzyme concentration also increases during the pregnancy. Acid phosphatase. This enzyme is found in highest concentration in the prostrate carcinoma. In Treatment of cancer A major potential therapeutic application of enzymes is in the treatment of cancer. Asparaginase has proved to be particularly promising for the treatment of acute lymphocytic leukemia. Its action depends upon the fact that tumour cells are deficient in aspartate-ammonia ligase activity, which restricts their ability to synthesise the normally non-essential amino acid L-asparagine. Therefore, they are forced to extract it from body fluids. The action of the asparaginase does not affect the functioning of normal cells which are able to synthesize enough for
their own requirements, but reduce the free exogenous concentration and so induces a state of fatal starvation in the susceptible tumour cells. A 60% incidence of complete remission has been reported in a study of almost 6000 cases of acute lymphocytic leukemia. The enzyme is administered intravenously. It is only effective in reducing asparagine levels within the bloodstream, showing a halflife of about a day (in a dog). This half-life may be increased 20-fold by use of polyethylene glycol-modified asparaginase. Enzyme deficiencies Phenylketonuria is detected by presence of phenylalanine in the urine. Some inborn error of metabolism is also detected are relatively harmless e.g. albinism , alkaptonuria, Table 8.1 showing List of defective enzyme and their diseases Alkaptonuria Phenylketonuria Maple syrup disease Galactosomia uridyltransferasae Glycogen storage Fructosuria Gaucher disease Tay Sachs Wilson disease Acetalasaemia Xerderma pigmentosa homogenistate 1 oxidase phenylalanine 4 monooxygenase oxo acid decorboxylase galactose-1 phosphate glucose 6 phosphate fructokinase glucocerbrosidase -NAcetykll D-hexosaminidase P-type ATPase catalase DNA binding protein helicases
Enzyme inhibitors and drug design Inhibition of hydroxymethylglutaryl Co-A reductase (HMGCo-A), during hypercholesterolemia. Other competitive inhibitors of HMGCo-A is namely mevinolin, compactin and monacol K which inhibits the enzyme. Inhibition of xanthin oxidase. Gout disease is characterize by uric acid that is end product of purine breakdown in humans derived from breakdown of nucleic acid. The main inhibitor is used allopurinol. Inhibition of HIV protease.The replication of HIV-1 is the cause of AIDS. The HIV1 protease catalyses this protein sequence. HIV protease is an aspartyl protease having a sequence Asp-thr-Gly at its active site. It resemble pepsin, and other retroviral protease. Many inhibitors have been designed that are analogous like rennin. Figure below showing structure of HIV protease. Figure 8. 1 HIV protease Use of enzyme in determination of concentration of metabolite of clinical importance. There are number of enzymatic advantage method estimate the metabolite in presence of many other substances an inhibitor present in serum may completely inhibit enzyme activity. For the estimation of certain metabolite e.g. glucose, and plasma glycerides enzyme methods are now used almost exclusively e.g. serum creatine and non-enzymic method are still being used in clinical chemistry. Blood glucose In the investigation of diabetes there are three enzymatic method that are available; hexokinase couple with glucose 6-phosphate dehydrogenase, glucose oxidase coupled with peroxidase. The second method is glucose oxidase is more commonly used. ________________________________________ Table 8.2 Some important therapeutic enzymes Enzyme EC number Reaction Use Asparaginase 3.5.1.1 L-Asparagine H2O L-aspartate + NH3 Leukaemia Collagenase 3.4.24.3 Collagen hydrolysis Skin ulcers Glutaminase 3.5.1.2 L-Glutamine H2O L-glutamate + NH3 Leukaemia Hyaluronidasea 3.2.1.35 Hyaluronate hydrolysis Heart attack Lysozyme 3.2.1.17 Bacterial cell wall hydrolysis Antibiotic Rhodanaseb 2.8.1.1 S2O32- + CN- SO32- + SCNCyanide poisoning Ribonuclease 3.1.26.4 RNA hydrolysis Antiviral -Lactamase 3.5.2.6 Penicillin penicilloate Penicillin allergy Streptokinasec 3.4.22.10 Plasminogen plasmin Blood clots Trypsin 3.4.21.4 Protein hydrolysis Inflammation Uricased 1.7.3.3 Urate + O2 allantoin Gout Urokinasee 3.4.21.31 Plasminogen plasmin Blood clots a Hyaluronoglucosaminidase, b thiosulphate sulfurtransferase,c streptococcal cysteine proteinase d urate oxidase,e plasminogen activator ________________________________________
Disadvantages of Using Enzyme as Therapeutic Agents As enzymes are specific biological catalysts, they should make the most desirable therapeutic agents for the treatment of metabolic diseases. Unfortunately a number of factors severely reduce this potential utility: a) They are too large to be distributed simply within the body's cells. This is the major reason why enzymes have not yet been successful applied to the large number of human genetic diseases. A number of methods are being developed in order to overcome this by targeting enzymes; as examples, enzymes with covalently attached external -galactose residues are targeted at hepatocytes and enzymes covalently coupled to target-specific monoclonal antibodies are being used to avoid non-specific side-reactions. b) Being generally foreign proteins to the body, they are antigenic and can elicit an immune response which may cause severe and life-threatening allergic reactions, particularly .on continued use. It has proved possible to circumvent this problem, in some cases, by disguising the enzyme as an apparently nonproteinaceous molecule by covalent modification. Asparaginase, modified by covalent attachment of polyethylene glycol, has been shown to retain its anti-tumour effect whilst possessing no immunogenicity. Clearly the presence of toxins, pyrogens and other harmful materials within a therapeutic enzyme preparation is totally forbidden. Effectively, this encourages the use of animal enzymes, in spite of their high cost, relative to those of microbial origin. Their effective lifetime within the circulation may be only a matter of minutes. This has proved easier than the immunological problem to combat, by disguise using covalent modification. Other methods have also been shown to be successful, particularly those involving entrapment of the enzyme within artificial liposomes, synthetic microspheres and red blood cell ghosts. However, although these methods are efficacious at extending the circulatory lifetime of the enzymes, they often cause increased immunological response and additionally may cause blood clots. In contrast to the industrial use of enzymes, therapeutically useful enzymes are required in relatively tiny amounts but at a very high degree of purity and (generally) specificity. The favoured kinetic properties of these enzymes are low Km and high Vmax in order to be maximally efficient even at very low enzyme and substrate concentrations. Thus the sources of such enzymes are chosen with care to avoid any possibility of unwanted contamination by incompatible material and to enable ready purification. Therapeutic enzyme preparations are generally offered for sale as lyophilised pure preparations with only biocompatible buffering salts and mannitol diluent added. The costs of such enzymes may be quite high but still comparable to those of competing therapeutic agents or treatments. As an example, urokinase (a serine protease, see Table 4.4) is prepared from human urine (some genetically engineered preparations are being developed) and used to dissolve blood clots. The cost of the enzyme is about Rs. 5000 mg-1, with the cost of treatment in a case of lung embolism being about Rs. 50000 for the enzyme alone. In spite of this, the market for the enzyme is worth about Rs 350M year-1. INDUSTRIAL ENZYME PROCESS AT HIGH TEMPERATURE 8.3 Enzyme operating temperature 0C Major applications alpha amylase (bacterial) 90-100 starch hydroysis, brewing baking detergents Gluco amylase 50-60 Maltodextrin hydrolysis alpha amylase (fungal) 50-60 Maltose Pullulanase 50-60 high glucose syrups Xylose isomerase 45-55 High fructose syrups Pectinase 20-50 clarification of juices /wines Cellulose 45-55 cellulose hydrolysis
Lactase food processing Acid protease fungal protease processing Alkaline proteases Lipases processing Enzyme used in Industries
30-50 30-50 40-60 40-60 30-70
lactose hydrolysis , food processing baking ,brewing,food Detergents Detergents, food
Thermozymes Thermozymes are thermostable enzymes that function optimally at 60 0C and 125 0C. these extremozymes are extremely valuable tools for study of protein stability Thermozymes are used in the molecular biology experiments for example taq polymerase and in the detergents (e.g.) proteases and starch processing (e.g. alpha amylase, glucsoe isomerases) various lipase and proteases oxidoreductase are used in the diagnostics, waste treatment and pulp and paper manufacture. Thermozymes are more stable because they are active against denaturing condition Hyperthermophiles have good potential for use in novel biotechnological processes including oil coal and waste gas desulphurization, heavy metal leaching and bioconversion of crude oils. Thermostable enzymes such as DNA polymerase, amylases, xylanases proteases and lipase are required in basic research and biotechnology. Organic solvent tolerant bacteria that exhibit the ability to degrade crude oil, polyaromatic hydrocarbons, or cholesterol or can utilize sulphure compounds have been isolated. e.g. Strain Y-40 degrades hydrocarbons and belongs to Candida. It can grow well in the presence of solvents such as n-octane, isooctane, cyclooctane, or kerosene at a concentration of 50 % (v/v) the first strain that isolated for organic solvent tolerant bacterium e.g. Pseudomonas putida can grow in more than 50% toluene. Obligate extremophiles such as thiobacillus thiooxidans and thiobacillus ferrooxidanse occur in highly acidic environments. T. ferroxidans reduces sulphure compounds as well as ferrous ion to ferric. This leads to production of acids. Currently these bacteria are utilized in the Biomining used in minerals leaching for example. T. ferroxidans used to isolate sulphure from the coal. Many extremophiles are used for craft industry for leather tanning (B. subtilis and B. licheniformis) with the help of various alkalophiles. Alkalophiles such as bacillus strains N-4 are used for cellulose digestion in the waste water treatment to digest cellulose. xylanases are produced by the alkalophiles Aeromonas strain 212 that can hydrolyze the beta 1.-4 linkage in the xylan polysaccharides in the crop plant. They show good activity and stability at pH 9. Neutral metalloproteases from B. amyloliquifaciens can be used in the brewing industry, when substituting malt with unmlated barley to increase the amount of free amino acids. The large-scale use of enzymes in solution Several enzymes, especially those used in starch processing, high-fructose syrup manufacture, textile desizing and detergent formulation, are now traded as commodity products on the world's markets. Although the cost of enzymes for use at the research scale is often very high, where there is a clear large-scale need for an enzyme its relative cost reduces dramatically with increased production. Relatively few enzymes, notably those in detergents, meat tenderizers and garden composting agents, are sold directly to the public. Most are used by industry to produce improved or novel products, to bypass long and involved chemical synthetic pathways or for use in the separation and purification of isomeric mixtures. Many of the most useful, but least-understood, uses of free enzymes are in the food industry. Here they are used, together with endogenous enzymes, to produce or process foodstuffs, which are only rarely substantially refined. Their action, however apparently straightforward, is complicated due to the effect that small
amounts of by-products or associated reaction products have on such subjective effects as taste, smell, colour and texture. The use of enzymes in the non-food (chemicals and pharmaceuticals) sector is relatively straightforward. Products are generally separated and purified and, therefore, they are not prone to the subtleties available to food products. Most such enzymic conversions benefit from the use of immobilised enzymes or biphasic systems The use of enzymes in detergents The use of enzymes in detergent formulations is now common in developed countries, with over half of all detergents presently available containing enzymes. In spite of the fact that the detergent industry is the largest single market for enzymes at 25 - 30% of total sales. details of the enzymes used and the ways in which they are used, have rarely been published. Dirt comes in many forms and includes proteins, starches and lipids. In addition, clothes that have been starched must be freed of the starch. Using detergents in water at high temperatures and with vigorous mixing, it is possible to remove most types of dirt but the cost of heating the water is high and lengthy mixing or beating will shorten the life of clothing and other materials. The use of enzymes allows lower temperatures to be employed and shorter periods of agitation are needed, often after a preliminary period of soaking. In general, enzyme detergents remove protein from clothes soiled with blood, milk, sweat, grass, etc. far more effectively than non-enzyme detergents. However, using modern bleaching and brightening agents, the difference between looking clean and being clean may be difficult to discern. At present only proteases and amylases are commonly used. Although a wide range of lipases is known, it is only very recently that lipases suitable for use in detergent preparations have been described. Detergent enzymes must be cost-effective and safe to use. Early attempts to use proteases foundered because of producers and users developing hypersensitivity. This was combatted by developing dust-free granulates (about 0.5 mm in diameter) in which the enzyme is incorporated into an inner core, containing inorganic salts (e.g. NaCI) and sugars as preservative, bound with reinforcing, fibres of carboxymethyl cellulose or similar protective colloid. This core is coated with inert waxy materials made from paraffin oil or polyethylene glycol plus various hydrophilic binders, which later disperse in the wash. This combination of materials both prevents dust formation and protects the enzymes against damage by other detergent components during storage. Enzymes are used in surprisingly small amounts in most detergent preparations, only 0.4 - 0.8% crude enzyme by weight (about 1% by cost). It follows that the ability to withstand the conditions of use is a more important criterion than extreme cheapness. Once released from its granulated form the enzyme must withstand anionic and non-ionic detergents, soaps, oxidants such as sodium perborate which generate hydrogen peroxide, optical brighteners and various lessreactive materials (Table 15.4), all at pH values between 8.0 and 10.5. Although one effect of incorporating enzymes is that lower washing temperatures may be employed with consequent savings in energy consumption, the enzymes must retain activity up to 60C. ________________________________________ Table 15.4 Compositions of an enzyme detergent Constituent Composition (%) Sodium tripolyphosphate (water softener, loosens dirt)a 38.0 Sodium alkane sulphonate (surfactant) 25.0 Sodium perborate tetrahydrate (oxidising agent)25.0 Soap (sodium alkane carboxylates) 3.0 Sodium sulphate (filler, water softener) 2.5 Sodium carboxymethyl cellulose (dirt-suspending agent) 1.6 Sodium metasilicate (binder, loosens dirt) 1.0 Bacillus protease (3% active) 0.8
Fluorescent brighteners 0.3 Foam-controlling agents Trace Perfume Trace Water to 100% a A recent trend is to reduce this phosphate content for environmental reasons. It may be replaced by sodium carbonate plus extra protease. ________________________________________ The enzymes used are all produced using species of Bacillus, mainly by just two companies. Novo Industri A/S produce and supply three proteases, Alcalase, from B. licheniformis, Esperase, from an alkalophilic strain of a B. licheniformis and Savinase, from an alkalophilic strain of B. amyloliquefaciens (often mistakenly attributed to B. subtilis). GistBrocades produce and supply Maxatase, from B. licheniformis. Alcalase and Maxatase (both mainly subtilisin) are recommended for use at 10-65C and pH 7-10.5. Savinase and Esperase may be used at up to pH 11 and 12, respectively. The -amylase supplied for detergent use is Termamyl, the enzyme from B. licheniformis which is also used in the production of glucose syrups. -Amylase is particularly useful in dish-washing and de-starching detergents. In addition to the granulated forms intended for use in detergent powders, liquid preparations in solution in water and slurries of the enzyme in a non-ionic surfactant are available for formulating in liquid 'spotting' concentrates, used for removing stubborn stains. Preparations containing both Termamyl and Alcalase are produced, Termamyl being sufficiently resistant to proteolysis to retain activity for long enough to fulfil its function. It should be noted that all the proteolytic enzymes described are fairly nonspecific serine endoproteases, giving preferred cleavage on the carboxyl side of hydrophobic amino acid residues but capable of hydrolysing most peptide links. They convert their substrates into small, readily soluble fragments which can be removed easily from fabrics. Only serine protease; may be used in detergent formulations: thiol proteases (e.g. papain) would be oxidised by the bleaching agents, and metalloproteases (e.g. thermolysin) would lose their metal cofactors due to complexing with the water softening agents or hydroxyl ions. The enzymes are supplied in forms (as described above) suitable for formulation by detergent manufacturers. Domestic users are familiar with powdered preparations but liquid preparations for home use are increasingly available. Household laundering presents problems quite different from those of industrial laundering: the household wash consists of a great variety of fabrics soiled with a range of materials and the user requires convenience and effectiveness with less consideration of the cost. Home detergents will probably include both an amylase and a protease and a lengthy warm-water soaking time will be recommended. Industrial laundering requires effectiveness at minimum cost so heated water will be re-used if possible. Large laundries can separate their 'wash' into categories and thus minimise the usage of water and maximise the effectiveness of the detergents. Thus white cotton uniforms from an abattoir can be segregated for washing, only protease being required. A pre-wash soaking for 10-20 min at pH up to 11 and 30-40C is followed by a main wash for 10-20 min at pH 11 and 60-65C. The water from these stages is discarded to the sewer. A third wash includes hypochlorite as bleach which would inactivate the enzymes rapidly. The water from this stage is used again for the pre-wash but, by then, the hypochlorite concentration is insufficient to harm the enzyme. This is essentially a batch process: hospital laundries may employ continuous washing machines, which transfer less-initially-dirty linen from a pre-rinse initial stage, at 32C and pH 8.5, into the first wash at 60C and pH 11, then to a second wash, containing hydrogen peroxide, at 71C and pH 11, then to a bleaching stage and rinsing. Apart from the pre-soak stage, from which water is run to waste, the process operates countercurrently. Enzymes are used in the pre-wash and in the first wash, the levels of peroxide at this stage being insufficient to inactivate the enzymes. There are opportunities to extend the use of enzymes in detergents both
geographically and numerically. They have not found widespread use in developing countries which are often hot and dusty, making frequent washing of clothes necessary. The recent availability of a suitable lipase may increase the quantities of enzymes employed very significantly. There are, perhaps, opportunities for enzymes such as glucose oxidase, lipoxygenase and glycerol oxidase as means of generating hydrogen peroxide in situ. Added peroxidases may aid the bleaching efficacy of this peroxide. A recent development in detergent enzymes has been the introduction of an alkaline-stable fungal cellulase preparation for use in washing cotton fabrics. During use, small fibres are raised from the surface of cotton thread, resulting in a change in the 'feel' of the fabric and, particularly, in the lowering of the brightness of colours. Treatment with cellulase removes the small fibres without apparently damaging the major fibres and restores the fabric to its 'as new' condition. The cellulase also aids the removal of soil particles from the wash by hydrolyzing associated cellulose fibres. 15.12 Applications of proteases in the food industry Certain proteases have been used in food processing for centuries and any record of the discovery of their activity has been lost in the mists of time. Rennet (mainly chymosin), obtained from the fourth stomach (abomasum) of unweaned calves has been used traditionally in the production of cheese. Similarly, papain from the leaves and unripe fruit of the pawpaw (Carica papaya) has been used to tenderize meats. These ancient discoveries have led to the development of various food applications for a wide range of available proteases from many sources, usually microbial. Proteases may be used at various pH values, and they may be highly specific in their choice of cleavable peptide links or quite non-specific. Proteolysis generally increases the solubility of proteins at their isoelectric points. 15.13 Rennet in cheese making The action of rennet in cheese making is an example of the hydrolysis of a specific peptide linkage, between phenylalanine and methionine residues (-Phe105Met106-) in the -casein protein present in milk . The -casein acts by stabilizing the colloidal nature of the milk, its hydrophobic N-terminal region associating with the lipophilic regions of the otherwise insoluble - and casein molecules, whilst its negatively charged C-terminal region associates with the water and prevents the casein micelles from growing too large. Hydrolysis of the labile peptide linkage between these two domains, resulting in the release of a hydrophilic glycosylated and phosphorylated oligopeptide (caseino macropeptide) and the hydrophobic para--casein, removes this protective effect, allowing coagulation of the milk to form curds, which are then compressed and turned into cheese (Figure 4.1). The coagulation process depends upon the presence of Ca2+ and is very temperature dependent (Q10 = 11) and so can be controlled easily. Calf rennet, consisting of mainly chymosin with a small but variable proportion of pepsin, is a relatively expensive enzyme and various attempts have been made to find cheaper alternatives from microbial sources These have ultimately proved to be successful and microbial rennets are used for about 70% of US cheese and 33% of cheese production world-wide. ________________________________________ Figure 15. 2 Outline method for the preparation of cheese. ________________________________________ The major problem that had to be overcome in the development of the microbial rennets was temperature lability. Chymosin is a relatively unstable enzyme and once it has done its major job, little activity remains. However, the enzyme from Mucor miehei retains activity during the maturation stages of cheese-making and produces bitter off-flavours. Treatment of the enzyme with oxidising agents (e.g. H2O2, peracids), which convert methionine residues to their sulfoxides, reduces its thermostability by about 10C and renders it more comparable with calf rennet. This is a rare example of enzyme technology being used to destabilise an enzyme
Attempts have been made to clone chymosin into Escherichia coli and Saccharomyces cerevisiae but, so far, the enzyme has been secreted in an active form only from the latter. The development of unwanted bitterness in ripening cheese is an example of the role of proteases in flavour production in foodstuffs. The action of endogenous proteases in meat after slaughter is complex but 'hanging' meat allows flavour to develop, in addition to tenderising it. It has been found that peptides with terminal acidic amino acid residues give meaty, appetising flavours akin to that of monosodium glutamate. Non-terminal hydrophobic amino acid residues in medium-sized oligopeptides give bitter flavours, the bitterness being less intense with smaller peptides and disappearing altogether with larger peptides. Application of this knowledge allows the tailoring of the flavour of protein hydrolysates. The presence of proteases during the ripening of cheeses is not totally undesirable and a protease from Bacillus amyloliquefaciens may be used to promote flavour production in cheddar cheese. Lipases from Mucor miehei or Aspergillus niger are sometimes used to give stronger flavours in Italian cheeses by a modest lipolysis, increasing the amount of free butyric acid. They are added to the milk (30 U l-1) before the addition of the rennet. When proteases are used to depolymerise proteins, usually non-specifically, the extent of hydrolysis (degree of hydrolysis) is described in DH units where: (15.1) Commercially, using enzymes such as subtilisin, DH values of up to 30 are produced using protein preparations of 8-12% (w/w). The enzymes are formulated so that the value of the enzyme : substrate ratio used is 2-4% (w/w). At the high pH needed for effective use of subtilisin, protons are released during the proteolysis and must be neutralised: subtilisin (pH 8.5) H2N-aa-aa-aa-aa-aa-COO- H2N-aa-aa-aa-COO- + H2N-aa-aa-COO- + H+ [15.2] where aa is an amino acid residue. Correctly applied proteolysis of inexpensive materials such as soya protein can increase the range and value of their usage, as indeed occurs naturally in the production of soy sauce. Partial hydrolysis of soya protein, to around 3.5 DH greatly increases its 'whipping expansion', further hydrolysis, to around 6 DH improves its emulsifying capacity. If their flavours are correct, soya protein hydrolysates may be added to cured meats. Hydrolysed proteins may develop properties that contribute to the elusive, but valuable, phenomenon of 'mouth feel' in soft drinks. Proteases are used to recover protein from parts of animals (and fish) would otherwise go to waste after butchering. About 5% of the meat can be removed mechanically from bone. To recover this, bones are mashed incubated at 60C with neutral or alkaline proteases for up to 4 h. The meat slurry produced is used in canned meats and soups. Large quantities of blood are available but, except in products such black puddings, they are not generally acceptable in foodstuffs because of their colour. The protein is of a high quality nutritionally and is de-haemed using subtilisin. Red cells are collected and haemolysed in water. Subtilisin is added and hydrolysis is allowed to proceed batchwise, with neutralization of the released protons, to around 18 DH, when the hydrophobic haem molecules precipitate. Excessive degradation is avoided to prevent the formation of bitter peptides. The enzyme is inactivated by a brief heat treatment at 85C and the product is centrifuged; no residual activity allowed into meat products. The haem-containing precipitate is recycled and the light-brown supernatant is processed through activated carbon beads to remove any residual haem. The purified hydrolysate, obtained in 60% yield, may be spray-dried and is used in cured meats, sausages and luncheon meats. 15.14 Meat tenderization Meat tenderisation is the softening of the meat for uses in the food by the endogenous proteases in the muscle after slaughter is a complex process which
varies with the nutritional, physiological and even psychological (i.e. frightened or not) state of the animal at the time of slaughter. Meat of older animals remains tough but can be tenderised by injecting inactive papain into the jugular vein of the live animals shortly before slaughter. Injection of the active enzyme would rapidly kill the animal in an unacceptably painful manner so the inactive oxidised disulfide form of the enzyme is used. On slaughter, the resultant reducing conditions cause free thiols to accumulate in the muscle, activating the papain and so tenderising the meat. This is a very effective process as only 2 - 5 ppm of the inactive enzyme needs to be injected. Recently, however, it has found disfavour as it destroys the animals heart, liver and kidneys that otherwise could be sold and, being reasonably heat stable, its action is difficult to control and persists into the cooking process. 15.15 Baking industry Proteases are also used in the baking industry. Where appropriate, dough may be prepared more quickly if its gluten is partially hydrolysed. A heat-labile fungal protease is used so that it is inactivated early in the subsequent baking. Weakgluten flour is required for biscuits in order that the dough can be spread thinly and retain decorative impressions. In the past this has been obtained from European domestic wheat but this is being replaced by high-gluten varieties of wheat. The gluten in the flour derived from these must be extensively degraded if such flour is to be used efficiently for making biscuits or for preventing shrinkage of commercial pie pastry away from their aluminium dishes. 15.16 The use of enzymes in starch hydrolysis Starch is the commonest storage carbohydrate in plants. It is used by the plants themselves, by microbes and by higher organisms so there is a great diversity of enzymes able to catalyse its hydrolysis. Starch from all plant sources occurs in the form of granules which differ markedly in size and physical characteristics from species to species. Chemical differences are less marked. The major difference is the ratio of amylose to amylopectin; e.g. corn starch from waxy maize contains only 2% amylose but that from amylomaize is about 80% amylose. Some starches, for instance from potato, contain covalently bound phosphate in small amounts (0.2% approximately), which has significant effects on the physical properties of the starch but does not interfere with its hydrolysis. Acid hydrolysis of starch has had widespread use in the past. It is now largely replaced by enzymic processes, as it required the use of corrosion resistant materials, gave rise to high colour and saltash content (after neutralisation), needed more energy for heating and was relatively difficult to control. ________________________________________ Figure 15. 3 The use of enzymes in processing starch. Typical conditions are given. ________________________________________ Of the two components of starch, amylopectine presents the great challenge to hydrolytic enzyme systems. This is due to the residues involved in -1,6glycosidic branch points which constitute about 4 - 6% of the glucose present. Most hydrolytic enzymes are specific for -1,4-glucosidic links yet the -1,6glucosidic links must also be cleaved for complete hydrolysis of amylopectin to glucose. Some of the most impressive recent exercises in the development of new enzymes have concerned debranching enzymes. It is necessary to hydrolyse starch in a wide variety of processes which may be condensed into two basic classes: Processes in which the starch hydrolysate is to be used by microbes or man, and Processes in which it is necessary to eliminate starch. In the former processes, such as glucose syrup production, starch is usually the major component of reaction mixtures, whereas in the latter processes, such as the processing of sugar cane juice, small amounts of starch which contaminate nonstarchy materials are removed. Enzymes of various types are used in these processes. Although starches from diverse plants may be utilised, corn is the
world's most abundant source and provides most of the substrate used in the preparation of starch hydrolysates. There are three stages in the conversion of starch (Figure 15.3): gelatinization, involving the dissolution of the nano-gram-sized starch granules to form a viscous suspension; liquefaction, involving the partial hydrolysis of the starch, with concomitant loss in viscosity; and Saccharification, involving the production of glucose and maltose by further hydrolysis. a. Gelatinization is achieved by heating starch with water, and occurs necessarily and naturally when starchy foods are cooked. Gelatinized starch is readily liquefied by partial hydrolysis with enzymes or acids and saccharified by further acidic or enzymic hydrolysis. The starch and glucose syrup industry uses the expression dextrose equivalent or DE, similar in definition to the DH units of proteolysis, to describe its products, where: (15 .3) In practice, this is usually determined analytically by use of the closely related, but not identical, expression: (15.4) Thus, DE represents the percentage hydrolysis of the glycosidic linkages present. Pure glucose has a DE of 100, pure maltose has a DE of about 50 (depending upon the analytical methods used; see equation (15.4)) and starch has a DE of effectively zero. During starch hydrolysis, DE indicates the extent to which the starch has been cleaved. Acid hydrolysis of starch has long been used to produce 'glucose syrups' and even crystalline glucose (dextrose monohydrate). Very considerable amounts of 42 DE syrups are produced using acid and are used in many applications in confectionery. Further hydrolysis using acid is not satisfactory because of undesirably coloured and flavoured breakdown products. Acid hydrolysis appears to be a totally random process which is not influenced by the presence of -1,6-glucosidic linkages. ________________________________________ Table 15.5 Enzymes used in starch hydrolysis Enzyme EC number Source Action -Amylase 3.2.1.1 Bacillus amyloliquefaciens Only -1,4-oligosaccharide links are cleaved to give -dextrins and predominantly maltose (G2), G3, G6 and G7 oligosaccharides B. licheniformis Only 1,4-oligosaccharide links are cleaved to give -dextrins and predominantly maltose, G3, G4 and G5 oligosaccharides Aspergillus oryzae, A. niger Only -1,4 oligosaccharide links are cleaved to give -dextrins and predominantly maltose and G3 oligosaccharides Saccharifying -amylase 3.2.1.1 B. subtilis (amylosacchariticus) Only -1,4-oligosaccharide links are cleaved to give -dextrins with maltose, G3, G4 and up to 50% (w/w) glucose -Amylase 3.2.1.2 Malted barley Only -1,4-links are cleaved, from nonreducing ends, to give limit dextrins and -maltose Glucoamylase 3.2.1.3 A. niger -1,4 and 1,6-links are cleaved, from the nonreducing ends, to give -glucose Pullulanase 3.2.1.41 B. acidopullulyticus Only -1,6-links are cleaved to give straight-chain maltodextrins ________________________________________ The nomenclature of the enzymes used commercially for starch hydrolysis The nomenclature of the enzymes used commercially for starch hydrolysis is somewhat confusing and the EC numbers sometimes lump together enzymes with subtly different activities (Table 15.5). For example, -amylase may be subclassified as liquefying or saccharifying amylases but even this classification is inadequate to encompass all the enzymes that are used in commercial starch hydrolysis. One reason for the confusion in the nomenclature is the use of the anomeric form of
the released reducing group in the product rather than that of the bond being hydrolysed; the products of bacterial and fungal -amylases are in the configuration and the products of -amylases are in the -configuration, although all these enzymes cleave between -1,4-linked glucose residues. The -amylases (1,4--D-glucan glucanohydrolases) are endohydrolases which cleave 1,4--D-glycosidic bonds and can bypass but cannot hydrolyze 1,6--D-glucosidic branch points. Commercial enzymes used for the industrial hydrolysis of starch are produced by Bacillus amyloliquefaciens (supplied by various manufacturers) and by B. licheniformis (supplied by Novo Industri A/S as Termamyl). They differ principally in their tolerance of high temperatures, Termamyl retaining more activity at up to 110C, in the presence of starch, than the B. amyloliquefaciens -amylase. The maximum DE obtainable using bacterial -amylases is around 40 but prolonged treatment leads to the formation of maltulose (4--D-glucopyranosyl-Dfructose), which is resistant to hydrolysis by glucoamylase and -amylases. DE values of 8-12 are used in most commercial processes where further saccharification is to occur. The principal requirement for liquefaction to this extent is to reduce the viscosity of the gelatinised starch to ease subsequent processing. Different approaches for starch liquefaction Various manufacturers use different approaches to starch liquefaction using amylases but the principles are the same. Granular starch is slurried at 30-40% (w/w) with cold water, at pH 6.0-6.5, containing 20-80 ppm Ca2+ (which stabilizes and activates the enzyme) and the enzyme is added (via a metering pump). The amylase is usually supplied at high activities so that the enzyme dose is 0.5-0.6 kg tonne-1 (about 1500 U kg-1 dry matter) of starch. When Termamyl is used, the slurry of starch plus enzyme is pumped continuously through a jet cooker, which is heated to 105C using live steam. Gelatinization occurs very rapidly and the enzymic activity, combined with the significant shear forces, begins the hydrolysis. The residence time in the jet cooker is very brief. The partly gelatinized starch is passed into a series of holding tubes maintained at 100105C and held for 5 min to complete the gelatinization process. Hydrolysis to the required DE is completed in holding tanks at 90-100C for 1 to 2 h. These tanks contain baffles to discourage backmixing. Similar processes may be used with B. amyloliquefaciens -amylase but the maximum temperature of 95C must not be exceeded. This has the drawback that a final 'cooking' stage must be introduced when the required DE has been attained in order to gelatinize the recalcitrant starch grains present in some types of starch which would otherwise cause cloudiness in solutions of the final product. Saccharification The liquefied starch is usually saccharified but comparatively small amounts are spray-dried for sale as 'maltodextrins' to the food industry mainly for use as bulking agents and in baby food. In this case, residual enzymic activity may be destroyed by lowering the pH towards the end of the heating period. Fungal -amylase also finds use in the baking industry. It often needs to be added to bread-making flours to promote adequate gas production and starch modification during fermentation. This has become necessary since the introduction of combine harvesters. They reduce the time between cutting and threshing of the wheat, which previously was sufficient to allow a limited sprouting so increasing the amounts of endogenous enzymes. The fungal enzymes are used rather than those from bacteria as their action is easier to control due to their relative heat lability, denaturing rapidly during baking. The liquefied starch at 8 -12 DE is suitable for saccharification to produce syrups with DE values of from 45 to 98 or more. The greatest quantities produced are the syrups with DE values of about 97. At present these are produced using the exoamylase, glucan 1,4--glucosidase (1,4--D-glucan glucohydrolase, commonly called glucoamylase but also called amyloglucosidase and -amylase), which releases -D-glucose from 1,4--, 1,6-- and 1,3--linked glucans. In theory, carefully liquefied starch at 8 -12 DE can be hydrolysed completely to produce a final glucoamylase reaction mixture with DE of 100 but, in
practice, this can be achieved only at comparatively low substrate concentrations. The cost of concentrating the product by evaporation decreases that a substrate concentration of 30% is used. It follows that the maximum DE attainable is 96 - 98 with syrup composition 95 - 97% glucose, 1 - 2% maltose and 0.5 - 2% (w/w) isomaltose (-D-glucopyranosyl-(1,6)-D-glucose). This material is used after concentration, directly for the production of high-fructose syrups or for the production of crystalline glucose. Whereas liquefaction is usually a continuous process, saccharification is most often conducted as a batch process. The glucoamylase most often used is produced by Aspergillus niger strains. This has a pH optimum of 4.0 - 4.5 and operates most effectively at 60C, so liquefied starch must be cooled and its pH adjusted before addition of the glucoamylase. The cooling must b rapid, to avoid retrogradation (the formation of intractable insoluble aggregates of amylose; the process that gives rise to the skin on custard). Any remaining bacterial -amylase will be inactivated when the pH is lowered; however, this may be replaced later by some acid-stable -amylase which is normally present in the glucoamylase preparations. When conditions are correct the glucoamylase is added, usually at the dosage of 0.65 - 0.80 litre enzyme preparation.tonne-1 starch (200 U kg-1). Saccharification is normally conducted in vast stirred tanks, which may take several hours to fill (and empty), so time will be wasted if the enzyme is added only when the reactors are full. The alternatives are to meter the enzyme at a fixed ratio or to add the whole dose of enzyme at the commencement of the filling stage. The latter should give the most economical use of the enzyme. ________________________________________ Figure 15. 4. The % glucose formed from 30% (w/w) 12 DE maltodextrin, at 60C and pH 4.3, using various enzyme solutions. 200 U kg-1 Aspergillus niger glucoamylase; -----------400 U kg-1 A. niger glucoamylase; 200 U kg-1 A. niger glucoamylase plus 200 U kg-1 Bacillus acidopullulyticus pullulanase. The relative improvement on the addition of pullulanase is even greater at higher substrate concentrations. ________________________________________ The saccharification process takes about 72 h to complete but may, of course, be speeded up by the use of more enzyme. Continuous saccharification is possible and practicable if at least six tanks are used in series. It is necessary to stop the reaction, by heating to 85C for 5 min, when a maximum DE has been attained. Further incubation will result in a fall in the DE, to about 90 DE eventually, caused by the formation of isomaltose as accumulated glucose re-polymerises with the approach of thermodynamic equilibrium (Figure 15.4). Final step in syrup formation The saccharified syrup is filtered to remove fat and denatured protein released from the starch granules and may then be purified by passage through activated charcoal and ion-exchange resins. It should be remembered that the dry substance concentration increases by about 11 % during saccharification, because one molecule of water is taken up for each glycosidic bond hydrolysed (molecule of glucose produced). Although glucoamylase catalyses the hydrolysis of 1,6--linkages, their breakdown is slow compared with that of 1,4--linkages (e.g. the rates of hydrolyzing the 1,4-, 1,6- and 1,3--links in tetrasaccharides are in the proportions 300 : 6 : 1). It is clear that the use of a debranching enzyme would speed the overall saccharification process but, for industrial use such an enzyme must be compatible with glucoamylase. Two types of debranching enzymes are available: pullulanase, which acts as an exo hydrolase on starch dextrins; and isoamylase (EC.3.2.1.68), which is a true endohydrolase. Novo Industri A/S have recently introduced a suitable pullulanase, produced by a strain of Bacillus acidopullulyticus. The pullulanase from Klebsiella aerogenes which has been available commercially to some time is unstable at temperatures over 45C but the B. acidopullulyticus enzymes can be used under the same conditions as the Aspergillus glucoamylase
(60C, pH 4.0-4.5). The practical advantage of using pullulanase together with glucoamylase is that less glucoamylase need be used This does not in itself give any cost advantage but because less glucoamylase is used and fewer branched oligosaccharides accumulate toward the end of the saccharification, the point at which isomaltose production becomes significant occurs at higher DE (Figure 4.3). It follows that higher DE values and glucose contents can be achieved when pullulanase is use (98 - 99 DE and 95 - 97% (w/w) glucose, rather than 97 - 98 DE) and higher substrate concentrations (30 - 40% dry solids rather than 25 - 30%) may be treated. The extra cost of using pullulanase is recouped by savings in evaporation and glucoamylase costs. In addition, when the product is to be used to manufacture high-fructose syrups, there is a saving in the cost of further processing. The development of the B. acidopullulyticus pullulanase is an excellent example of what can be done if sufficient commercial pull exists for a new enzyme. The development of a suitable -D-glucosidase, in order to reduce the reversion, would be an equally useful step for industrial glucose production. Screening of new strains of bacteria for a novel enzyme of this type is a major undertaking. It is not surprising that more details of the screening procedures used are not readily available. 15.17 Production of syrups containing maltose Traditionally, syrups containing maltose as a major component have been produced by treating barley starch with barley -amylase. -Amylases (1,4--D-glucan maltohydrolases) are exohydrolases which release maltose from 1,4--linked glucans but neither bypass nor hydrolyse 1,6--linkages. High-maltose syrups (40 - 50 DE, 45-60% (w/w) maltose, 2 - 7% (w/w) glucose) tend not to crystallise, even below 0C and are relatively non-hygroscopic. They are used for the production of hard candy and frozen deserts. High conversion syrups (60 - 70 DE, 30 - 37% maltose, 35 - 43% glucose, 10% maltotriose, 15% other oligosaccharides, all by weight) resist crystallisation above 4C and are sweeter (Table 4.3). They are used for soft candy and in the baking, brewing and soft drinks industries. It might be expected that -amylase would be used to produce maltose-rich syrups from corn starch, especially as the combined action of -amylase and pullulanase give almost quantitative yields of maltose. This is not done on a significant scale nowadays because presently available -amylases are relatively expensive, not sufficiently temperature stable (although some thermostable -amylases from species of Clostridium have recently been reported) and are easily inhibited by copper and other heavy metal ions. Instead fungal -amylases, characterised by their ability to hydrolyse maltotriose (G3) rather than maltose (G2) are employed often in combination with glucoamylase. Presently available enzymes, however, are not totally compatible; fungal -amylases requiring a pH of not less than 5.0 and a reaction temperature not exceeding 55C. High-maltose syrups (see Figure 15.3) are produced from liquefied starch of around 11 DE at a concentration of 35% dry solids using fungal -amylase alone. Saccharification occurs over 48 h, by which time the fungal -amylase has lost its activity. Now that a good pullulanase is available, it is possible to use this in combination with fungal -High-conversion syrups are produced using combinations of fungal -amylase and glucoamylase. These may be tailored to customers' specifications by adjusting the activities of the two enzymes used but inevitably, as glucoamylase is employed, the glucose content of the final product will be higher than that of high-maltose syrups. The stability of glucoamylase necessitates stopping the reaction, by heating, when the required composition is reached. It is now possible to produce starch hydrolysates with any DE between 1 and 100 and with virtually any composition using combinations of bacterial amylases, fungal -amylases, glucoamylase and pullulanase. ________________________________________ Table 15.6 The relative sweetness of food ingredients Food ingredient Relative sweetness (by weight, solids) Sucrose 1.0
Glucose 0.7 Fructose 1.3 Galactose 0.7 Maltose 0.3 Lactose 0.2 Raffinose 0.2 Hydrolysed sucrose 1.1 Hydrolysed lactose 0.7 Glucose syrup 11 DE <0.1 Glucose syrup 42 DE 0.3 Glucose syrup 97 DE 0.7 Maltose syrup 44 DE 0.3 High-conversion syrup 65 DE 0.5 HFCS (42% fructose)a 1.0 HFCS (55% fructose) 1.1 Aspartame 180 a HFCS, high-fructose corn syrup. 15.18 Enzymes in the sucrose industry The sucrose industry is a comparatively minor user of enzymes but provides few historically significant and instructive examples of enzyme technology The hydrolysis ('inversion') of sucrose, completely or partially, to glucose and fructose provides sweet syrups that are more stable (i.e. less likely crystallise) than pure sucrose syrups. The most familiar 'Golden Syrup' produced by acid hydrolysis of one of the less pure streams from the cane sugar refinery but other types of syrup are produced using yeast (Saccharomyces cerevisiae) invertase. Although this enzyme is unusual in that it suffers from substrate inhibition at high sucrose levels ( > 20% (w/w)), this does not prevent its commercial use at even higher concentrations: Figure 15. 5Traditionally, Invertase was produced on site by autolysing yeast cells. The autolysate was added to the syrup (70% sucrose (w/w)) to be inverted together with small amounts of xylene to prevent microbial growth. Inversion was complete in 48 - 72 h at 50C and pH 4.5. The enzyme and xylene were removed during the subsequent refining and evaporation. Partially inverted syrups were (and still are) produced by blending totally inverted syrups with sucrose syrups. Now, commercially produced invertase concentrates are employed. The production of hydrolysates of a low molecular weight compound in essentially pure solution seems an obvious opportunity for the use of an immobilised enzyme, yet this is not done on a significant scale, probably because of the extreme simplicity of using the enzyme in solution and the basic conservatism of the sugar industry. Invertase finds another use in the production of confectionery with liquid or soft centres. These centers are formulated using crystalline sucrose and tiny (about 100 U kg-1, 0.3 ppm (w/w)) amounts of invertase. At this level of enzyme, inversion of sucrose is very slow so the centre remains solid long enough for enrobing with chocolate to be completed. Then, over a period of days or weeks, sucrose hydrolysis occurs and the increase in solubility causes the centers to become soft or liquid, depending on the water content of the centre preparation. Other enzymes are used as aids to sugar production and refining by removing materials which inhibit crystallisation or cause high viscosity. In some parts of the world, sugar cane contains significant amounts of starch, which becomes viscous, thus slowing filtration processes and making the solution hazy when the sucrose is dissolved. This problem can be overcome by using the most thermostable -amylases (e.g. Termamyl at about 5 U kg-1) which are entirely compatible with the high temperatures and pH values that prevail during the initial vacuum
evaporation stage of sugar production. Other problems involving dextran and raffinose required the development of new industrial enzymes. A Dextran is produced by the action of dextransucrase (EC 2.4.1.5) from Leuconostoc mesenteroides on sucrose and found as a slime on damaged cane and beet tissue, especially when processing has been delayed in hot and humid climates. Raffinose, which consists of sucrose with -galactose attached through its C-1 atom to the 6 position on the glucose residue, is produced at low temperatures in sugar beet. Both dextran and raffinose have the sucrose molecule as part of their structure and both inhibit sucrose crystal growth. This produces plate-like or needle-like crystals which are not readily harvested by equipment designed for the approximately cubic crystals otherwise obtained. Dextran can produce extreme viscosity it process streams and even bring plant to a stop. Extreme dextran problems arc frequently solved by the use of fungal dextranases produced from Penicillium species. These are used (e.g. 10 U kg-1 raw juice, 55C, pH 5.5, 1 h) only in times of crisis as they are not sufficiently resistant to thermal denaturation for long-term use and are inactive at high sucrose concentrations. Because only small quantities are produced for use, this enzyme is relatively expensive. An enzyme sufficiently stable for prophylactic use would be required in order to benefit from economies of scale. Raffinose may be hydrolysed to galactose and sucrose by a fungal raffinase. 15.19 Glucose from cellulose There is very much more cellulose available, as a potential source of glucose, than starch, yet cellulose is not a significant source of pure glucose. The reasons for this are many, some technical, some commercial. The fundamental reason is that starch is produced in relatively pure forms by plants for use as an easily biodegradable energy and carbon store. Cellulose is structural and is purposefully combined and associated with lignin and pentosans, so as to resist biodegradation; dead trees take several years to decay even in tropical rainforests. A typical waste cellulolytic material contains less than half cellulose, most of the remainder consisting of roughly equal quantities of lignin and pentosans. A combination of enzymes is needed to degrade this mixture. These enzymes are comparatively unstable of low activity against native lignocellulose and subject to both substrate and product inhibition. Consequently, although many cellulolytic enzymes exist and it is possible to convert pure cellulose to glucose using only enzymes, the cost of this conversion is excessive. The enzymes might be improved by strain selection from the wild or by mutation but problems caused by the physical nature of cellulose are not so amenable to solution. Granular starch is readily stirred in slurries containing 40% (w/v) solids and is easily solubilised but, even when pure, fibrous cellulose forms immovable cakes at 10% solids and remains insoluble in all but the most exotic (and enzyme denaturing) solvents. Impure cellulose often contains almost an equal mass of lignin, which is of little or no value as a by-product and is difficult an expensive to remove. Commercial cellulase preparations from Trichoderma reesei consist of mixtures of the synergistic enzymes: cellulase (EC 3.2.1.4), an endo-1,4-D -glucanase; glucan 1,4--glucosidase (EC 3.2.1.74), and exo-1,4--glucosidase; and cellulose 1,4--cellobiosidase (EC 3.2.1.91), an exo-cellobiohydrolase (see Figure 15.6). They are used for the removal of relatively small concentrations of cellulose complexes which have been found to interfere in the processing of plant material in, for example, the brewing and fruit juice industries. ________________________________________ Figure 15. 6 Outline of the relationship between the enzyme activities in the hydrolysis of cellulose. || represents inhibitory effects. Endo-1,4--glucanase is the rate-controlling activity and may consist of a mixture of enzymes acting on cellulose of different degrees of crystallinity. It acts synergistically with both
exo-1,4--glucosidase and exo-cellobiohydrolase. Exo-1,4--glucosidase is a product-inhibited enzyme. Exo-cellobiohydrolase is product inhibited and additionally appears to be inactivated on binding to the surface of crystalline cellulose. ________________________________________ Proper economic analysis reveals that cheap sources of cellulose prove to be generally more expensive as sources of glucose than apparently more expensive starch. Relatively pure cellulose is valuable in its own right, as a paper pulp and chipboard raw material, which currently commands a price of over twice that of corn starch. With the increasing world shortage of pulp it cannot be seen realistically as an alternative source of glucose in the foreseeable future. Knowledge of enzyme systems capable of degrading lignocellulose is advancing rapidly but it is unlikely that lignocellulose will replace starch as a source of glucose syrups for food use. It is, however, quite possible that it may be used, in a process involving the simultaneous use of both enzymes and fermentative yeasts, to produce ethanol; the utilisation of the glucose by the yeast removing its inhibitory effect on the enzymes. It should be noted that cellobiose is a nonfermentable sugar and must be hydrolysed by additional -glucosidase (EC 3.2.1.21, also called cellobiase for maximum process efficiency (Figure 15.6). 15.20 The use of lactases in the dairy industry Lactose is present at concentrations of about 4.7% (w/v) in milk and the whey (supernatant) left after the coagulation stage of cheese-making. Its presence in milk makes it unsuitable for the majority of the world's adult population, particularly in those areas which have traditionally not had a dairy industry. Real lactose tolerance is confined mainly to peoples whose origins lie in Northern Europe or the Indian subcontinent and is due to 'lactase persistence'; the young of all mammals clearly are able to digest milk but in most cases this ability reduces after weaning. Of the Thai, Chinese and Black American populations, 97%, 90% and 73% respectively, are reported to be lactose intolerant, whereas 84% and 96% of the US White and Swedish populations, respectively, are tolerant. Additionally, and only very rarely some individuals suffer from inborn metabolic lactose intolerance or lactase deficiency, both of which may be noticed at birth. The need for low-lactose milk is particularly important in food-aid programmes as severe tissue dehydration, diarrhoea and even death may result from feeding lactose containing milk to lactose-intolerant children and adults suffering from protein-calorie malnutrition. In all these cases, hydrolysis of the lactose to glucose and galactose would prevent the (severe) digestive problems. Another problem presented by lactose is its low solubility resulting in crystal formation at concentrations above 11 % (w/v) (4C). This prevents the use of concentrated whey syrups in many food processes as they have a unpleasant sandy texture and are readily prone to microbiological spoilage. Adding to this problem, the disposal of such waste whey is expensive (often punitively so) due to its high biological oxygen demand. These problems may be overcome by hydrolysis of the lactose in whey; the product being about four times as sweet (see Table 15.6), much more soluble and capable of forming concentrated, microbiologically secure, syrups (70% (w/v)). Figure 15. 7 Lactose may be hydrolysed by lactase, a -galactosidase. Commercially, it may be prepared from the dairy yeast Kluyveromyces fragilis (K. marxianus var. marxianus), with a pH optimum (pH 6.5-7.0) suitable for the treatment of milk, or from the fungi Aspergillus oryzae or A. niger, with pH optima (pH 4.5-6.0 and 3.0-4.0, respectively) more suited to whey hydrolysis. These enzymes are subject to varying degrees of product inhibition by galactose. In addition, at high lactose and galactose concentrations, lactase shows significant transferase ability and produces -1,6-linked galactosyl
oligosaccharides. Lactases are now used in the production of ice cream and sweetened flavoured and condensed milks. When added to milk or liquid whey (2000 U kg-1) and left for about a day at 5C about 50% of the lactose is hydrolysed, giving a sweeter product which will not crystallise if condensed or frozen. This method enables otherwise-wasted whey to replace some or all of the skim milk powder used in traditional ice cream recipes. It also improves the 'scoopability' and creaminess of the product. Smaller amounts of lactase may be added to long-life sterilised milk to produce a relatively inexpensive lactose-reduced product (e.g. 20 U kg-1, 20C, 1 month of storage). Generally, however, lactase usage has not reached its full potential, as present enzymes are relatively expensive and can only be used at low temperatures. 15.21 Enzymes in the fruit juice, wine, brewing and distilling industries One of the major problems in the preparation of fruit juices and wine is cloudiness due primarily to the presence of pectins. These consist primarily of 1,4-anhydrogalacturonic acid polymers, with varying degrees of methyl esterification. They are associated with other plant polymers and, after homogenisation, with the cell debris. The cloudiness that they cause is difficult to remove except by enzymic hydrolysis. Such treatment also has the additional benefits of reducing the solution viscosity, increasing the volume of juice produced (e.g. the yield of juice from white grapes can be raised by 15%), subtle but generally beneficial changes in the flavour and, in the case of wine-making, shorter fermentation times. Insoluble plant material is easily removed by filtration, or settling and decantation, once the stabilising effect of the pectins on the colloidal haze has been removed. Commercial pectolytic enzyme preparations are produced from Aspergillus niger and consist of a synergistic mixture of enzymes: polygalacturonase (EC 3.2.1.15), responsible for the random hydrolysis of 1,4- D-galactosiduronic linkages; pectinesterase (EC 3.2.1.11), which releases methanol from the pectyl methyl esters, a necessary stage before the polygalacturonase can act fully (the increase in the methanol content of such treated juice is generally less than the natural concentrations and poses no health risk); pectin lyase (EC 4.2.2.10), which cleaves the pectin, by an elimination reaction releasing oligosaccharides with non-reducing terminal 4-deoxymethyl--D-galact-4enuronosyl residues, without the necessity of pectin methyl esterase action; and hemicellulase (a mixture of hydrolytic enzymes including: xylan endo-1,3-xylosidase, EC 3.2.1.32; xylan 1,4--xylosidase, EC 3.2.1.37; and -Larabinofuranosidase, EC 3.2.1.55), strictly not a pectinase but its adventitious presence is encouraged in order to reduce hemicellulose levels. The optimal activity of these enzymes is at a pH between 4 and 5 and generally below 50C. They are suitable for direct addition to the fruit pulps at levels around 20 U l-1 (net activity). Enzymes with improved characteristics of greater heat stability and lower pH optimum are currently being sought. In brewing, barley malt supplies the major proportion of the enzyme needed for saccharification prior to fermentation. Often other starch containing material (adjuncts) are used to increase the fermentable sugar and reduce the relative costs of the fermentation. Although malt enzyme may also be used to hydrolyse these adjuncts, for maximum economic return extra enzymes are added to achieve their rapid saccharification. It not necessary nor desirable to saccharify the starch totally, as non-fermentable dextrins are needed to give the drink 'body' and stabilise its foam 'head'. For this reason the saccharification process is stopped, by boiling the 'wort', after about 75% of the starch has been converted into fermentable sugar. The enzymes used in brewing are needed for saccharification of starch (bacterial and fungal -amylases), breakdown of barley -1,4- and -1,3- linked glucan (glucanase) and hydrolysis of protein (neutral protease) to increase the (later)
fermentation rate, particularly in the production of high-gravity beer, where extra protein is added. Cellulases are also occasionally used, particularly where wheat is used as adjunct but also to help breakdown the barley -glucans. Due to the extreme heat stability of the B. amyloliquefaciens -amylase, where this is used the wort must be boiled for a much longer period (e.g. 30 min) to inactivate it prior to fermentation. Papain is used in the later post-fermentation stages of beer-making to prevent the occurrence of protein- and tannin-containing 'chillhaze' otherwise formed on cooling the beer. Recently, 'light' beers, of lower calorific content, have become more popular. These require a higher degree of saccharification at lower starch concentrations to reduce the alcohol and total solids contents of the beer. This may be achieved by the use of glucoamylase and/or fungal -amylase during the fermentation. A great variety of carbohydrate sources are used world wide to produce distilled alcoholic drinks. Many of these contain sufficient quantities of fermentable sugar (e.g. rum from molasses and brandy from grapes), others contain mainly starch and must be saccharified before use (e.g. whiskey from barley malt, corn or rye). In the distilling industry, saccharification continues throughout the fermentation period. In some cases (e.g. Scotch malt whisky manufacture uses barley malt exclusively) the enzymes are naturally present but in others (e.g. grain spirits production) the more heat-stable bacterial -amylases may be used in the saccharification. 15.22 Glucose oxidase and catalase in the food industry Glucose oxidase is a highly specific enzyme, from the fungi Aspergillus niger and Penicillium, which catalyses the oxidation of -glucose to glucono-1,5-lactone (which spontaneously hydrolyses non-enzymically to gluconic acid) using molecular oxygen and releasing hydrogen peroxide. It finds uses in the removal of either glucose or oxygen from foodstuffs in order to improve their storage capability. Hydrogen peroxide is an effective bacteriocide and may be removed, after use, by treatment with catalase (derived from the same fungal fermentations as the glucose oxidase) which converts it to water and molecular oxygen: catalase 2H2O2 2H2O + O2 [4.4] For most large-scale applications the two enzymic activities are not separated. Glucose oxidase and catalase may be used together when net hydrogen peroxide production is to be avoided. A major application of the glucose oxidase/catalase system is in the removal of glucose from egg-white before drying for use in the baking industry. A mixture of the enzymes is used (165 U kg-1) together with additional hydrogen peroxide (about 0.1 % (w/w)) to ensure that sufficient molecular oxygen is available, by catalase action, to oxidise the glucose. Other uses are in the removal of oxygen from the head-space above bottled and canned drinks and reducing non-enzymic browning in wines and mayonnaises.
Chapter-16
Enzyme Reactors
Introduction An enzyme reactor consists of a vessel, or series of vessels, used to perform a desired conversion by enzymic means. There are several important factors that determine the choice of reactor for a particular process. In general, the choice depends on the cost of a predetermined productivity within the product's specifications. This must be inclusive of the costs associated with substrate(s), downstream processing, labour, depreciation, overheads and process development, in addition to the more obvious costs concerned with building and running the enzyme reactor. Other contributing factors are the form of the enzyme of choice (i.e. free or immobilised), the kinetics of the reaction and the chemical and physical properties of an immobilization support including whether it is particulate, membranous or fibrous, and its density, compressibility, robustness, particle size and regenerability. Attention must also be paid to the scale of operation, the possible need for pH and temperature control, the supply and removal of gases and the stability of the enzyme, substrate and product. These factors will be discussed in more detail with respect to the different types of reactor. There are two type of Reactor 1. Batch Reactor; 2. Continuous reactor Stirred tank batch reactor (STR). Batch membrane reactor (MR). Packed bed reactor (PBR). continuous flow stirred tank reactor (CSTR. continuous flow membrane reactor (CMR) fluidised bed reactor (FBR) 16.1 Batch Reactor Batch reactors generally consist of a tank containing a stirrer (stirred tank reactor, STR). Note that a batch reactor is one in which all of the product is removed, as rapidly as is practically possible, after a fixed time. The tank is normally fitted with fixed baffles that improve the stirring efficiency. Generally this means that the enzyme and substrate molecules must have identical residence times within the reactor, although in some circumstances there may be a need for further additions of enzyme and/or substrate (i.e. fed -batch operation). a. Disadvantage of using batch reactor 1. The operating costs of batch reactors are higher than for continuous processes due to the necessity for the reactors to be emptied and refilled both regularly and often. This procedure is not only expensive in itself but means that there are considerable periods when such reactors are not productive; it also makes uneven demands on both labour and services. 2. STRs can be used for processes involving non-immobilised enzymes, if the consequences of these contaminating the product are not severe. 3. Batch reactors also suffer from pronounced batch-to-batch variations, as the
reaction conditions change with time, and may be difficult to scale-up, due to the changing power requirements for efficient fixing. b. Advantage of using Batch Bioreactor They do, however, have a number of advantageous features. Primary amongst these is their simplicity both in use and in process development. For this reason they are preferred for small-scale production of highly priced products, especially where the same equipment is to be used for a number of different conversions. They offer a closely controllable environment that is useful for slow reactions, where the composition may be accurately monitored, and conditions (e.g. temperature, pH, coenzyme concentrations) varied throughout the reaction. They are also of use when continuous operation of a process proves to be difficult due to the viscous or intractable nature of the reaction mix. ________________________________________ Figure 16. 1 Diagrams of various important enzyme reactor types. Stirred tank batch reactor (STR), which contains all of the enzyme and substrates) until the conversion is complete; batch membrane reactor (MR), where the enzyme is held within membrane tubes which allow the substrate to diffuse in and the product to diffuse out. This reactor may often be used in a semicontinuous manner, using the same enzyme solution for several batches; packed bed reactor (PBR), also called plug -flow reactor (PFR), containing a settled bed of immobilised enzyme particles; continuous flow stirred tank reactor (CSTR) which is a continuously operated version of (a); continuous flow membrane reactor (CMR) which is a continuously operated version of (b); fluidised bed reactor (FBR), where the flow of gas and/or substrate keeps the immobilised enzyme particles in a fluidised state. All reactors would additionally have heating/cooling coils (interior in reactors (a), and (d), and exterior, generally, in reactors (b), (c), (e) and (f)) and the stirred reactors may contain baffles in order to increase (reactors (a), (b), (d) and (e) or decrease (reactor (f)) the stirring efficiency. The continuous reactors ((c) -(f)) may all be used in a recycle mode where some, or most, of the product stream is mixed with the incoming substrate stream. All reactors may use immobilised enzymes. In addition, reactors (a), (b) and (e) (plus reactors (d) and (f), if semipermeable membranes are used on their outlets) may be used with the soluble enzyme. ________________________________________ c. The expected productivity of a batch reactor The expected productivity of a batch reactor may be calculated by, assuming the validity of the non -reversible Michaelis -Menten reaction scheme with no diffusional control, inhibition or denaturation. The rate of reaction (v) may be expressed in terms of the volume of substrate solution within the reactor (VolS) and the time (t): (16.1) Therefore: (16.2) On integrating using the boundary condition that [S] = [S]0 at time (t) = 0: (16.3) Let the fractional conversion be X, where: (16.4) Therefore; (16.4a) and (16.4b) Also
(16.4c) Therefore substituting using (5.4c) and (5.4b) in (5.3): (16.5) The change in fractional conversion and concentrations of substrate and product with time in a batch reactor is shown in Figure 16.2(a).
Figure 16. 2 This figure shows two related behaviours. (a) The change in substrate and product concentrations with time, in a batch reactor. The reaction S P is assumed, with the initial condition [S]0/Km = 10. The concentrations of substrate ( and product (-----------) are both normalised with respect to [S]0. The normalised time (i.e. t = t Vmax/[S]0) is relative to the time (t = 1) that would be required to convert all the substrate if the enzyme acted at Vmax throughout, the actual time for complete conversion being longer due to the reduction in the substrate concentration at the reaction progresses. The dashed line also indicates the variation of the fractional conversion (X) with t. (b) The change in substrate and product concentrations with reactor length for a PBR. The reaction S P is assumed with the initial condition, [S]0/Km = 10. The concentrations of substrate () and product (-----------) are both normalised with respect to [S]". The normalised reactor length (i.e. I = lVmax/F, where Vmax is the maximum velocity for unit reactor length and I is the reactor length) is relative to the length (i.e. when I = 1) that contains sufficient enzyme to convert all the substrate at the given flow rate if the enzyme acted at its maximum velocity throughout; the actual reactor length necessary for complete conversion being longer due to the reduction in the substrate concentration as the reaction progresses. P may be considered as the relative position within a PBR or the reactor's absolute length. ________________________________________ 16.2 Membrane reactors The main requirement for a membrane reactor (MR) is a semipermeable membrane which allows the free passage of the product molecules but contains the enzyme molecules. A cheap example of such a membrane is the dialysis membrane used for removing low molecular weight species from protein preparations. The usual choice for a membrane reactor is a hollow-fibre reactor consisting of a preformed module containing hundreds of thin tubular fibres each having a diameter of about 200 m and a membrane thickness of about 50 m. Advantage of using Membrane reactor [1] Membrane reactors may be used in either batch or continuous mode and [2] they allow the easy separation of the enzyme from the product. [3] They are normally used with soluble enzymes, avoiding the costs and problems associated with other methods of immobilization and some of the diffusion limitations of immobilized enzymes. If the substrate is able to diffuse through the membrane, it may be introduced to either side of the membrane with respect to the enzyme; otherwise it must be within the same compartment as the enzyme, a configuration that imposes a severe restriction on the flow rate through the reactor, if used in continuous mode.[4] Due to the ease with which membrane reactor systems may be established, they are often used for production on a small scale (g to kg), especially where a multi-enzyme pathway or coenzyme regeneration is needed.[5] They allow the easy replacement of the enzyme in processes involving particularly labile enzymes and can also be used for biphasic reactions. The major disadvantage of these reactors concerns the cost of the membranes and their need to be replaced at regular intervals. 16.3 The kinetics of membrane reactors The kinetics of membrane reactors are similar to those of the batch STR, in batch mode, or the CSTR, in continuous mode (see later). Deviations from these models occur primarily in configurations where the substrate stream is on the side of the membrane opposite to the enzyme and the reaction is severely limited by its
diffusion through the membrane and the products' diffusion in the reverse direction. Under these circumstances the reaction may be even more severely affected by product inhibition or the limitations of reversibility than is indicated by these models. 16.4 Packed bed reactors (PBR) The most important characteristic of a PBR is that; they are also called plug flow reactors (PFR) because material flows through the reactor as a plug. Ideally, all of the substrate stream flows at the same velocity, parallel to the reactor axis with no back -mixing. All material present at any given reactor cross -section has had an identical residence time. The longitudinal position within the PBR is, therefore, proportional to the time spent within the reactor; all product emerging with the same residence time and all substrate molecule having an equal opportunity for reaction. The conversion efficiency of a PBR, with respect to its length, behaves in a manner similar to that of a well -stirred batch reactor with respect to its reaction time (Figure 5.2(b)) Each volume element behaves as a batch reactor as it passes through the PBR. Any required degree of reaction may be achieved by use of an idea PBR of suitable length. The flow rate (F) is equivalent to VolS/t for a batch reactor. Therefore equation (16.5) may be converted to represent an ideal PBR, given the assumption, not often realised in practice, that there are no diffusion limitations: (16.6) In order to produce ideal plug -flow within PBRs, a turbulent flow regime is preferred to laminar flow, as this causes improved mixing and heat transfer normal to the flow and reduced axial back-mixing. Achievement of high enough Re may, however, be difficult due to unacceptably high feed rates. Consequent upon the plug -flow characteristic of the PBR is that the substrate concentration is maximised, and the product concentration minimised, relative to the final conversion at every point within the reactor; the effectiveness factor being high on entry to the reactor and low close to the exit. This means that PBRs are the preferred reactors, all other factors being equal, for processes involving product inhibition, substrate activation and reaction reversibility. At low Re the flow rate is proportional to the pressure drop across the PBR. This pressure drop is, in turn, generally found to be proportional to the bed height, the linear flow rate and dynamic viscosity of the substrate stream and (1 - )2/3 (where is the porosity of the reactor; i.e. the fraction of the PBR volume taken up by the liquid phase), but inversely proportional to the cross-sectional area of the immobilised enzyme pellets. In general PBRs are used with fairly rigid immobilised-enzyme catalysts (1 -3 mm diameter), because excessive increases in this flow rate may distort compressible or physically weak particles. Particle deformation results in reduced catalytic surface area of particles contacting the substrate-containing solution, poor external mass transfer characteristics and a restriction to the flow, causing increased pressure drop. A vicious circle of increased back-pressure, particle deformation and restricted flow may eventually result in no flow at all through the PBR. PBRs behave as deep-bed filters with respect to the substrate stream. It is necessary to use a guard bed if plugging of the reactor by small particles is more rapid than the biocatalysts' deactivation. They are also easily fouled by colloidal or precipitating material. The design of PBRs does not allow for control of pH, by addition of acids or bases, or for easy temperature control where there is excessive heat output, a problem that may be particularly noticeable in wide reactors (> 15 cm diameter). Deviations from ideal plug-flow are due to back-mixing within the reactors, the resulting product streams having a distribution of residence times. In an extreme case, back-mixing may result in the kinetic behaviour of the reactor approximating to that of the CSTR (see below), and the consequent difficulty in achieving a high degree of conversion. These deviations are caused by channeling, where some substrate passes through the reactor more rapidly, and hold-up, which involves stagnant areas with negligible flow rate. Channels may form in the reactor bed due
to excessive pressure drop, irregular packing or uneven application of the substrate stream, causing flow rate differences across the bed. The use of a uniformly sized catalyst in a reactor with an upwardly flowing substrate stream reduces the chance and severity of non-ideal behavior. 16.5 Continuous flow stirred tank reactors (CSTIR) This reactor consists of a well -stirred tank containing the enzyme, which is normally immobilised. The substrate stream is continuously pumped into the reactor at the same time as the product stream is removed. This is the example of continous reactor. If the reactor is behaving in an ideal manner, there is total back-mixing and the product stream is identical with the liquid phase within the reactor and invariant with respect to time. Some molecules of substrate may be removed rapidly from the reactor, whereas others may remain for substantial periods. The distribution of residence times for molecules in the substrate stream is shown in Figure 16.2 Advantages The CSTR is an easily constructed, versatile and cheap reactor, which allows simple catalyst charging and replacement. Its well -mixed nature permits straightforward control over the temperature and pH of the reaction and the supply or removal of gases. CSTRs tend to be rather large as the: need to be efficiently mixed. Their volumes are usually about five to ten time the volume of the contained immobilised enzyme. This, however, has the advantage that there is very little resistance to the flow of the substrate stream, which may contain colloidal or insoluble substrates, so long as the insoluble particles are not able to sweep the immobilised enzyme from the reactor. The mechanical nature of the stirring limits the supports for the immobilised enzymes to materials which do not easily disintegrate to give 'fines' which may enter the product stream. However, fairly small particle (down to about 10 m diameter) may be used, if they are sufficiently dense to stay within the reactor. This minimises problems due to diffusional resistance. An ideal CSTR has complete back -mixing resulting in a minimisation of the substrate concentration, and a maximisation of the product concentration, relative to the final conversion, at every point within the reactor the effectiveness factor being uniform throughout. Thus, CSTRs are the preferred reactors, everything else being equal, for processes involving substrate inhibition or product activation. They are also useful where the substrate stream contains an enzyme inhibitor, as it is diluted within the reactor. This effect is most noticeable if the inhibitor concentration is greater than the inhibition constant and [S]0/Km is low for competitive inhibition or high for uncompetitive inhibition, when the inhibitor dilution has more effect than the substrate dilution. Deviations from ideal CSTR behaviour occur when there is a less effective mixing regime and may generally be overcome by increasing the stirrer speed, decreasing the solution viscosity or biocatalyst concentration or by more effective reactor baffling. Kinetics of CSTR The rate of reaction within a CSTR can be derived from a simple mass balance to be the flow rate (F) times the difference in substrate concentration between the reactor inlet and outlet. Hence: (16.7) Therefore: (16.8) from equation (5.4): (16.9) Therefore: (16.10) This equation should be compared with that for the PBR (equation (16.6)). Together these equations can be used for comparing the productivities of the two reactors
(Figures 16.4 and 16.5). ________________________________________ Figure 16. 3Figure 5.4. The residence time distribution of a CSTR. The relative number of molecules resident within the reactor for a particular time N, is plotted against the normalised residence time (i.e. t F/V, where V is the reactor volume, and F is the flow rate; it is the time relative to that required for one reactor volume to pass through the reactor). The residence time distribution of non -reacting media molecules ( ----------- which obeys the relationship , where [M] is the concentration of media molecules, giving a half-life for remaining in the reactor of , product ( ) and substrate () are shown. The reaction S P is assumed, and substrate molecules that have long residence times are converted into product, the average residence time of the product being greater than that for the substrate. The composition of the product stream is identical with that of the liquid phase within the reactor. This composition may be calculated from the relative areas under the curves and, in this case, represents a 90% conversion. Under continuous operating conditions (operating time > 4V/F), the mean residence time within the reactor is V/F. However, it may be noted from the graph that only a few molecules have a residence time close to this value (only 7% between 0.9V/F and 1.1 V/F) whereas 20% of the molecules have residence times of less than 0.1 V/F or greater than 2.3V/F. It should be noted that 100% of the molecules in an equivalent ideal PBR might be expected to have residence times equal to their mean residence time. ________________________________________ Figure 16. 4Figure 5.5. Comparison of the changes in fractional conversion with flow rate between the PBR () and CSTR (-----------) at different values of [S]0/Km (10,1 and 0.1, higher [S]0/Km giving the higher curves). The flow rate is normalised with respect to the reactor's volumetric enzyme content ( = FKm/Vmax. It can be seen that there is little difference between the two reactors at faster flow rates and lower conversions, especially at high values of [S]0/Km. ________________________________________ Figure 16. 5 Figure 5.6. Comparison of the changes in fractional conversion with residence time between the PBR () and CSTR (-----------) at different values of [S]0/Km (10, 1 and 0.1; higher [S]0/Km values giving the lower curves). The residence time is the reciprocal of the normalised flow rate (see Figure 5.5). If the flow rate is unchanged then the 'normalised residence time' may be thought of as the reactor volume needed to produce the required degree of conversion. ________________________________________ Equations describing the behaviour of CSTRs and PBRs utilising reversible reactions or undergoing product or substrate inhibition can be derived in a similar manner, using equations (1.68), (1.85) and (1.96) rather than (1.8): a. Substrate -inhibited PBR (16.11) Substrate -inhibited CSTR (16.12) b. Product -inhibited PBR (16.13) Product -inhibited CSTR (16.14) Reversible reaction in a PBR (16.15) Reversible reaction in a CSTR (16.16) X in equations (5.15) and (5.16) is the fractional conversion for a reversible reaction. (16.17)
These equations may be used to compare the size of PBR and CSTR necessary to achieve the same conversion under various conditions (Figure 16.6). The productivity Another useful parameter for comparing these reactors is the productivity. This can be derived for each reactor assuming a first-order inactivation of the enzyme (equation (1.26)). Combined with equation (5.6) for PBR, or (5.10) for CSTR, the following relationships are obtained on integration: PBR (16.18) CSTR (16.19)
where kd is the first -order inactivation constant (i.e. kd1, in equation (1.25)) and the fractional conversion subscripts refer to time = 0 or t. The change in productivity (Figure 16.7) and fractional conversion (Figure 16.8) of these reactors with time can be compared using these equations. These reactors may be operated for considerably longer periods than that determined by the inactivation of their contained immobilised enzyme, particularly if they are capable of high conversion at low substrate concentrations (Figure 16.8). This is independent of any enzyme stabilisation and is simply due to such reactors initially containing large amounts of redundant enzyme. ________________________________________ Figure 16. 6Figure 5.7. Comparison of the ratio, of the enzyme content in a CSTR to that in a PBR, necessary to achieve various degrees of conversion for a range of process conditions. The actual size of the CSTR will be five to ten times greater than indicated due to the necessity of maintaining stirring within the vessel. Uninhibited reaction; product inhibited; --------substrate inhibited. (curve a) [S]0/Km = 100; (curve b) [S]0/Km = 1; (curve c) [S]0/Km < 0.01; (curve d) [S]0/Km = 1, product inhibited KP/Km = 0.1 ; (curve e) [S]0/Km = 1, product inhibited KP/Km = 0.01; (curve f) [S]0/Km = 1, substrate inhibited [S]0/KS = 10; (curve g) [S]0/Km = 100; substrate inhibited [S]0/KS = 10. The size of a CSTR becomes prohibitively large at high conversions (e.g. using curve b, a CSTR contains three times the enzyme in a PBR to achieve a 90% conversion, but this increases to 18 times for 99% conversion. The difference between the two types of reactor is increased if the effectiveness factor () is less than one due to diffusional effects. ________________________________________ Figure 16. 7Figure 5.8. The change in productivity of a PBR (---------) and CSTR () with time, assuming an initial fractional conversion (X0) = 0.99 and [S]0/Km = 100. The units of time are half -lives of the free enzyme ( ) and the productivity is given in terms of (FXt1/2). Although the overall productivity is 1.6 times greater for a CSTR than a PBR, it should be noted that the CSTR contains 1.9 times more enzyme. ________________________________________ In general, there is little or no back -pressure to increased flow rate through the CSTR. Such reactors may be started up as batch reactors until the required degree of conversion is reached, when the process may be made continuous. CSTRs are not generally used in processes involving high conversions but a chain of CSTRs may approach the PBR performance. This chain may be a number (greater than three) of reactors connected in series or a single vessel divided into compartments, in order to minimise back-mixing CSTRs may be used with soluble rather than immobilised enzyme if an ultrafiltration membrane is used to separate the reactor output stream from the reactor contents. This causes a number of process difficulties, including concentration polarization or inactivation of the enzyme on the membrane but may be preferable in order to achieve a combined reaction and separation process or where a suitable immobilised enzyme is not
readily available. ________________________________________ Figure 16. 8Figure 5.9. The change in fractional conversion of PBRs and CSTRs with time, assuming initial fractional conversion (X0) of 0.99 or 0.80. CSTR, [S]o/Km = 0.01; - - - - - - CSTR, [S]0/Km = 100; PBR [S]0/Km = 0.01; ------- PBR [S]0/Km = 100. The time is given in terms of the half -life of the free enzyme ( ). Although the CSTR maintains its fractional conversion for a longer period than the PBR, particularly at high X0. It should be noted that a CSTR capable of X0 = 0.99 at a substrate feed concentration of [S]0/Km = 0.01 contains 22 times more enzyme than an equivalent PBR, but yields only 2.2 times more product. The initial stability in the fractional conversion over a considerable period of time, due to the enzyme redundancy, should not be confused with any effect due to stabilisation of the immobilised enzyme. 5. Fluidised bed reactors These reactors generally behave in a manner intermediate between CSTRs and PBRs. They consist of a bed of immobilised enzyme which is fluidised by the rapid upwards flow of the substrate stream alone or in combination with a gas or secondary liquid stream, either of which may be inert or contain material relevant to the reaction. A gas stream is usually preferred as it does not dilute the product stream. There is a minimum fluidisation velocity needed to achieve bed expansion, which depends upon the size, shape, porosity and density of the particles and the density and viscosity of the liquid. This minimum fluidisation velocity is generally fairly low (about 0.2 -I.0 cm s-1) as most immobilisedenzyme particles have densities close to that of the bulk liquid. In this case the relative bed expansion is proportional to the superficial gas velocity and inversely proportional to the square root of the reactor diameter. Fluidising the bed requires a large power input but, once fluidised, there is little further energetic input needed to increase the flow rate of the substrate stream through the reactor (Figure 16.2). At high flow rates and low reactor diameters almost ideal plug -flow characteristics may be achieved. However, the kinetic performance of the FBR normally lies between that of the PBR and the CSTR, as the small fluid linear velocities allowed by most biocatalytic particles causes a degree of backmixing that is often substantial, although never total. The actual design of the FBR will determine whether it behaves in a manner that is closer to that of a PBR or CSTR (see Figures 5.5 -5.9). It can, for example, be made to behave in a manner very similar to that of a PBR, if it is baffled in such a way that substantial backmixing is avoided. FBRs are chosen when these intermediate characteristics are required, e.g. where a high conversion is needed but the substrate stream is colloidal or the reaction produces a substantial pH change or heat output. They are particularly useful if the reaction involves the utilisation or release of gaseous material. The FBR is normally used with fairly small immobilised enzyme particles (20-40 m diameter) in order to achieve a high catalytic surface area. These particles must be sufficiently dense, relative to the substrate stream, that they are not swept out of the reactor. Less-dense particles must be somewhat larger. For efficient operation the particles should be of nearly uniform size otherwise a non-uniform biocatalytic concentration gradient will be formed up the reactor. FBRs are usually tapered outwards at the exit to allow for a wide range of flow rates. Very high flow rates are avoided as they cause channelling and catalyst loss. The major disadvantage of development of FBR process is the difficulty in scalingup these reactors. PBRs allow scale-up factors of greater than 50000 but, because of the markedly different fluidisation characteristics of different sized reactors, FBRs can only be scaled-up by a factor of 10 -100 each time. In addition, changes in the flow rate of the substrate stream causes complex changes in the flow pattern within these reactors that may have consequent unexpected effects upon the conversion rate.
Chapter-17
Protein Engineering
Introduction Protein engineering is the branch of science in which novel enzymes are being designed and produce by genetic engineering methods to improve the stability of an enzyme (stability with respect to chemical oxidation, temperature, and pH). Therefore protein engineering includes two parts one large scale production of genes by modification in the gene by gene cloning methods and other modification in the structure of protein to improve the substrate specificity, the cofactor requirement (NADPH to NADH) or imparting novel properties to a protein (eg. adding a Mn binding site into horse heart myoglobin at UBC).In the genetic engineering methods the availability of a cloned gene is important. Cloned gene is expressed in the expression vector to obtain the 3-D structure of the enzyme, resolved by Xray crystallography. In the absence of detailed 3-D structure, one can do random mutagenesis and/or directed evolution. There are many directions in which enzyme technologists are currently applying their art and which are at the forefront of biotechnological research and development. At present, relatively few enzymes are available on a large scale (i.e. > kg) and are suitable for industrial applications. These shortcomings are being addressed in a number of ways: New enzymes are being sought in the natural environment and by strain selection. Established industrial enzymes are being used in as wide a variety of ways as can be conceived. novel enzymes are being designed and produce by genetic engineering; New organic catalysts are being designed and synthesized. More complex enzyme systems are being utilised. The development of genetically improved enzymes is generally undertaken by molecular biologists and the design and synthesis of novel enzyme-like catalysts is done by the organic chemists. Both groups of workers will, however, base their science on data provided by the enzyme technologist. Space requirements in this volume do not allow the full treatment of these related areas but will be discussed briefly here. 17.1 Enzyme engineering A most exciting development over the last few years is the application of genetic engineering techniques to enzyme technology. There are a number of properties which may be improved or altered by genetic engineering for example, the yield and kinetics of the enzyme, the ease of downstream processing and various safety aspects. Enzymes from dangerous or unapproved microorganisms and from slow growing or limited plant or animal tissue may be cloned into safe high-production microorganisms. In the future, enzymes may be redesigned to fit more appropriately into industrial processes; for example, making glucose isomerase less susceptible to inhibition by the Ca2+ present in the starch saccharification processing stream. 17.2 Enzyme production can be increased by cloning of gene The amount of enzyme produced by a microorganism may be increased by increasing the number of gene copies that code for it by inserting a portion of gene for specific enzyme into the suitable vector. If the vector is expression type then large amount of protein will be obtained after the translation of vector. The
product can be tested by its specific function. The DNA having gene of interest for specific enzyme can be isolated by partial digestion of gene or complete digestion of gene (incomplete or complete fragmentation of gene) and selection of specific gene can be done with the help of probe. This principle has been used to increase the activity of penicillin-G-amidase in Escherichia coli. The cellular DNA from a producing strain is selectively cleaved by the restriction endonuclease Hind III. This hydrolyses the DNA at relatively rare sites containing the 5'-AAGCTT-3' base sequence to give identical 'staggered' ends. Figure 17. 1 figure showing staggered cut of DNA double strand by the restriction endonuclease. Note that single stranded DNA is rarely cut by the Restriction enzyme.
R.E. Intact DNA
cleaved DNA
Figure 17. 2 showing structure of vector and their site containing antibiotic resistance gene as the marker of the gene for selection of recombinant gene.
The total DNA is cleaved into about 10000 fragments, only one of which contains the required genetic information. These fragments are individual cloned into a cosmid vector and thereby returned to E. coli. These colonies containing the active gene are identified by their inhibition of a 6-amino-penicillanic acidsensitive organism. Such colonies are isolated and the penicillin-G-amidase gene transferred on to pBR322 plasmids and recloned back into E. coli. The engineered cells, aided by the plasmid amplification at around 50 copies per cell, produce penicillin-G-amidase constitutively and in considerably higher quantities than does the fully induced parental strain. Such increased yields are economically relevant not just for the increased volumetric productivity but also because of reduced downstream processing costs, the resulting crude enzyme being that much purer. Another extremely promising area of genetic engineering is protein engineering. New enzyme structures may be designed and produced in order to improve on existing enzymes or create new activities. An outline of the process of protein engineering is shown in Figure 17.1. Such factitious enzymes are produced by site-directed mutagenesis (Figure 17.2). Unfortunately from a practical point of view, much of the research effort in protein engineering has gone into studies concerning the structure and activity of enzymes chosen for their theoretical importance or ease of preparation rather than industrial relevance. This emphasis is likely to change in the future. Figure 17. 3 showing structure and location of plasmid DNA inside the E.coli vector. Figure 17. 4 showing nature of cutting of gene one sticky end Figure 17. 5 showing selection of recombinant by use antibiotic
As indicated by the method used for site-directed mutagenesis , the preferred pathway for creating new enzymes is by the stepwise substitution of only one or two amino acid residues out of the total protein structure. Although a large database of sequence-structure correlations is available, (NCBI) and growing rapidly together with the necessary software, it is presently insufficient
accurately to predict three-dimensional changes as a result of such substitutions. The main problem is assessing the long-range effects, including solvent interactions, on the new structure. As the many reported results would attest, the science is at a stage where it can explain the structural consequences of amino acid substitutions after they have been determined but cannot accurately predict them.
Figure 17. 6 showing complete procedure of gene cloning.
Figure 17. 7 The protein engineering cycle. The process starts with the isolation and characterization of the required enzyme. This information is analyzed together with the database of known and putative structural effects of amino acid substitutions to produce a possible improved structure. This factitious enzyme is constructed by site-directed mutagenesis, isolated and characterised. The results, successful or unsuccessful, are added to the database, and the process repeated until the required result is obtained. ________________________________________ Protein engineering, therefore, is presently rather a hit or miss process which may be used with only little realistic likelihood of immediate success. Apparently quite small sequence changes may give rise to large conformational alterations and even affect the rate-determining step in the enzymic catalysis. However it is reasonable to suppose that, given a sufficiently detailed database plus suitable software, the relative probability of success will increase over the coming years and the products of protein engineering will make a major impact on enzyme technology. 17.2 Application of Enzyme Engineering 17.2.1 Subtilisin Much protein engineering has been directed at subtilisin (from Bacillus amyloliquefaciens), the principal enzyme in the detergent enzyme preparation, Alcalase. This has been aimed at the improvement of its activity in detergents by stabilising it at even higher temperatures, pH and oxidant strength. Most of the attempted improvements have concerned alterations to: The P1 cleft, which holds the amino acid on the carbonyl side of the targeted peptide bond; 2. The oxyanion hole (principally Asn155), which stabilises the tetrahedral intermediate; 3. the neighborhood of the catalytic histidyl residue (His64), which has a general base role; and 4.the methionine residue (Met222) which causes subtilisin's lability to oxidation. It has been found that the effect of a substitution in the P1 cleft on the relative specific activity between substrates may be fairly accurately predicted even though predictions of the absolute effects of such changes are less successful. Many substitutions, particularly for the glycine residue at the bottom of the P1 cleft (Gly166), have been found to increase the specificity of the enzyme for particular peptide links whilst reducing it for others. These effects are achieved mainly by corresponding changes in the Km rather than the Vmax. Increases in relative specificity may be useful for some applications. They should not be thought of as the usual result of engineering enzymes, however, as native subtilisin is unusual in being fairly non-specific in its actions, possessing a large hydrophobic binding site which may be made more specific relatively easily (e.g. by reducing its size). The inactivation of subtilisin in bleaching solutions coincides with the conversion of Met222 to its sulfoxide, the consequential increase in volume occluding the oxyanion hole. Substitution of this methionine by serine or alanine produces mutants that are relatively stable, although possessing somewhat reduced activity. ________________________________________
Figure 17. 8 Figure 17. 9 An outline of the process of site-directed mutagenesis, using a hypothetical example. (a) The primary structure of the enzyme is derived from the DNA sequence. A putative enzyme primary structure is proposed with an asparagine residue replacing the serine present in the native enzyme. A short piece of DNA (the primer), complementary to a section of the gene apart from the base mismatch, is synthesised. (b) The oligonucleotide primer is annealed to a single-stranded copy of the gene and is extended with enzymes and nucleotide triphosphates to give a double-stranded gene. On reproduction, the gene gives rise to both mutant and wild-type clones. The mutant DNA may be identified by hybridisation with radioactively labelled oligonucleotides of complementary structure. ________________________________________ Trypsin An example of the unpredictable nature of protein engineering is given by trypsin, which has an active site closely related to that of subtilisin. Substitution of the negatively charged aspartic acid residue at the bottom of its P1 cleft (Asp189), which is used for binding the basic side-chains of lysine or arginine, by positively charged lysine gives the predictable result of abolishing the activity against its normal substrates but unpredictably also gives no activity against substrates where these basic residues are replaced by aspartic acid or glutamic acid. Thermophilic enzymes Considerable effort has been spent on engineering more thermophilic enzymes. It has been found that thermophilic enzymes are generally only 20-30 kJ more stable than their mesophilic counterparts. This may be achieved by the addition of just a few extra hydrogen bonds, an internal salt link or extra internal hydrophobic residues, giving a slightly more hydrophobic core. All of these changes are small enough to be achieved by protein engineering. To ensure a more predictable outcome, the secondary structure of the enzyme must be conserved and this generally restricts changes in the exterior surface of the enzyme. Suitable for exterior substitutions for increasing thermostability have been found to be aspartate < glutamate lysine, glutamine, valine < threonine, serine < asparagine, isoleucine < threonine, asparagine <aspartate and lysine < arginine. Such substitutions have a fair probability of success. Whenever there is small increases in the interior hydrophobicity, for example by substituting interior glycine or serine residues by alanine may also increase the thermostability. It should be recognized that making an enzyme more thermostable reduces its overall flexibility and, hence, it is probable that the factitious enzyme produced will have reduced catalytic efficiency. Artificial enzymes Synzymes A number of possibilities now exist for the construction of artificial enzymes. These are generally synthetic polymers or oligomers with enzyme-like activities, often called synzymes. They must possess two structural entities, a substratebinding site and a catalytically effective site. It has been found that producing the facility for substrate binding is relatively straightforward but catalytic sites are somewhat more difficult. Both sites may be designed separately but it appears that, if the synzyme has a binding site for the reaction transition state, this often achieves both functions. Synzymes generally obey the saturation Michaelis-Menten kinetics. For a one-substrate reaction the reaction sequence is given by synzyme + S (synzyme-S complex) synzyme + P [17.5] Some synzymes are simply derivatised proteins, although covalently immobilised enzymes are not considered here. An example is the derivatisation of myoglobin,
the oxygen carrier in muscle, by attaching (Ru(NH3)5)3+ to three surface histidine residues. This converts it from an oxygen carrier to an oxidase, oxidising ascorbic acid whilst reducing molecular oxygen. The synzyme is almost as effective as natural ascorbate oxidases. Disadvantage of synzymes It is impossible to design protein synzymes from scratch with any probability of success, as their conformations are not presently predictable from their primary structure. Such proteins will also show the drawbacks of natural enzymes, being sensitive to denaturation, oxidation and hydrolysis. For example, polylysine binds anionic dyes but only 10% as strongly as the natural binding protein, serum albumin, in spite of the many charges and apolar side-chains. Polyglutamic acid, however, shows synzymic properties. It acts as an esterase in much the same fashion as the acid proteases, showing a bell-shaped pH-activity relationship, with optimum activity at about pH 5.3, and Michaelis-Menten kinetics with a Km of 2 mm and Vmax of 10-4 to 10-5 s-1 for the hydrolysis of 4-nitrophenyl acetate. Cyclodextrins (Schardinger dextrins) are naturally occurring toroidal molecules consisting of six, seven, eight, nine or ten -1, 4-linked D-glucose units joined head-to-tail in a ring and -cyclodextrins, respectively: they may be synthesized from starch by the cyclomaltodextrin glucanotransferase (EC 2.4.1.19) from Bacillus macerans). They differ in the diameter of their cavities (about 0.5-1 nm) but all are about 0.7 nm deep. These form hydrophobic pockets due to the glycosidic oxygen atoms and inwards-facing C-H groups. All the C-6 hydroxyl groups project to one end and all the C-2 and C-3 hydroxyl groups to the other. Their overall characteristic is hydrophilic, being water soluble, but the presence of their hydrophobic pocket enables them to bind hydrophobic molecules of the appropriate size. Synzymic cyclodextrins are usually derivatised in order to introduce catalytically relevant groups. Many such derivatives have been examined. For example, a C-6 hydroxyl group of -cyclodextrin was covalently derivatised by an activated pyridoxal coenzyme. The resulting synzyme not only acted a transaminase (see reaction scheme [1.2]) but also showed stereoselectivity for the L-amino acids. It was not as active as natural transaminases, however. Polyethylenimine is formed by polymerising ethyleneimine to give a highly branched hydrophilic three-dimensional matrix. About 25% of the resultant amines are primary, 50% secondary and 25% tertiary: [17.6] Ethyleneimine polyethyleneimine The primary amines may be alkylated to form a number of derivatives. If 40% of them are alkylated with 1-iodododecane to give hydrophobic binding sites and the remainder alkylated with 4(5)-chloromethylimidazole to give general acid-base catalytic sites, the resultant synzyme has 27% of the activity of -chymotrypsin against 4-nitrophenyl esters. As might be expected from its apparently random structure, it has very low esterase specificity. Other synzymes may be created in a similar manner. Antibodies to transition state analogues of the required reaction may act as synzymes. For example, phosphonate esters of general formula (R-PO2-OR')- are stable analogues of the transition state occurring in carboxylic ester hydrolysis. Monoclonal antibodies raised to immunizing protein conjugates covalently attached to these phosphonate esters act as esterases. The specificities of these catalytic antibodies (also called abzymes) depends on the structure of the side-chains (i.e. R and R' in (R-PO2-OR')-) of the antigens. The Km values may be quite low, often in the micromolar region, whereas the Vmax values are low (below 1 s-1), although still 1000-fold higher than hydrolysis by background hydroxyl ions. A similar strategy may be used to produce synzymes by molecular 'imprinting' of polymers, using the presence of transition state analogues to shape polymerising resins or inactive non-enzymic protein during heat denaturation. 17.3 Advantages of genetic engineering
Many enzymes are now produced by fermentation of genetically modified microrganisms (GMOs). There are several advantages of using GMOs for the production of enzymes, including. It is possible to produce enzymes with a higher specificity and purity. It is possible to obtain enzymes which would otherwise not be available for economical, occupational health or environmental reasons. Due to higher production efficiency there is an additional environmental benefit through reducing energy consumption and waste from the production plants For enzymes used in the food industry particular benefits are for example a better use of raw materials (juice industry), better keeping quality of a final food and thereby less wastage of food (baking industry) and a reduced use of chemicals in the production process (starch industry). For enzymes used in the feed industry particular benefits include a significant reduction in the amount of phosphorus released to the environment from farming. The enzymes are produced by fermentation of the genetically modified microrganisms (the production strain), which then produces the desired enzyme. The process takes place under well controlled conditions in closed fermentation tank. After fermentation the enzyme is separated from the production strain, purified and mixed with inert diluents for stabilisation. Genetic engineering can be used to improve production and purification of recombinant enzymes, eg. targeting enzymes for extracellular secretion to allow for their easy isolation and purification. Examples of industrially useful enzymes include: A. proteases: subtilisin (detergent enzyme), rennet (cheese-processing). B. carbohydrases: amylases (starch hydrolysis), cellulases (bioconversions by Stake Technology), xylanases (bioconversions and biopulping), glucose isomerase (HFCS) C. pharmaceutically important enzymes: urokinase (plasminogen activator) is a protease for the treatment of thrombosis and embolism (Prve et al. 1987). The Lilly Research Labs hold the patent on the production of urokinase from a continuous line of porcine kidney cells D. ligninases and Mn peroxidases: pulp processing. E. restriction endonucleases: used extensively in research.
Chapter-18
Biosensors and Immunosensors
Introduction A biosensor is an analytical device which converts a biological response into an electrical signal (Figure 19.1). The term 'biosensor' is often used to cover sensor devices used in order to determine the concentration of substances. Clark and Lyons first demonstrated the modern concept of biosensors, in which an enzyme was immobilized into an electrode to form a biosensor. When the biological component of biosensor is a component of the immune system then it is referred to as Immunosensors. The emphasis of this Chapter concerns enzymes as the biologically responsive material, but it should be recognised that other biological systems may be utilised by biosensors, for example, whole cell metabolism, ligand binding and the antibody-antigen reaction. 19.1 The use of enzymes in analysis Enzymes make excellent analytical reagents due to their specificity, selectivity and efficiency. They are often used to determine the concentration of their substrates (as analytes) by means of the resultant initial reaction rates. If the reaction conditions and enzyme concentrations are kept constant, these rates of reaction (v) are proportional to the substrate concentrations ([S]) at low substrate concentrations. When [S] < 0.1 Km, equation from Michaelis velocity
constant simplifies to give v = (Vmax/Km)[S] The rates of reaction are commonly determined from the difference in optical absorbance between the reactants and products. An example of this is the -Dgalactose dehydrogenase (EC 1.1.1.48) assay for galactose which involves the oxidation of galactose by the redox coenzyme, nicotine-adenine dinucleotide (NAD+). -D-galactose + NAD+ D-galactono-1,4-lactone + NADH + H+ [19.1] A 0.1 mM solution of NADH has an absorbance at 340nm, in a 1 cm path-length cuvette, of 0.622, whereas the NAD+ from which it is derived has effectively zero absorbance at this wavelength. The conversion (NAD+ NADH) is, therefore, accompanied by a large increase in absorption of light at this wavelength. For the reaction to be linear with respect to the galactose concentration, the galactose is kept within a concentration range well below the Km of the enzyme for galactose. In contrast, the NAD+ concentration is kept within a concentration range well above the Km of the enzyme for NAD+, in order to avoid limiting the reaction rate. Such assays are commonly used in analytical laboratories and are, indeed, excellent where a wide variety of analyses need to be undertaken on a relatively small number of samples. The drawbacks to this type of analysis become apparent when a large number of repetitive assays need to be performed. Then, they are seen to be costly in terms of expensive enzyme and coenzyme usage, time consuming, labour intensive and in need of skilled and reproducible operation within properly equipped analytical laboratories. For routine or on-site operation, these disadvantages must be overcome. This is being achieved by the production of biosensors which exploit biological systems in association with advances in micro-electronic technology. Research and development in this field is wide and multidisciplinary, spanning biochemistry, bioreactor science, physical chemistry, electrochemistry, electronics and software engineering. Most of this current endeavour concerns potentiometric and amperometric biosensors and colorimetric paper enzyme strips. 19.2 A successful biosensor must possess at least some of the following beneficial features The biocatalyst must be highly specific for the purpose of the analyses, be stable under normal storage conditions and, except in the case of colorimetric enzyme strips and dipsticks (see later), show good stability over a large number of assays (i.e. much greater than 100). The reaction should be as independent of such physical parameters as stirring, pH and temperature as is manageable. This would allow the analysis of samples with minimal pre-treatment. If the reaction involves cofactors or coenzymes these should, preferably, also be co-immobilised with the enzyme. The response should be accurate, precise, reproducible and linear over the useful analytical range, without dilution or concentration. It should also be free from electrical noise. If the biosensor is to be used for invasive monitoring in clinical situations, the probe must be tiny and biocompatible, having no toxic or antigenic effects. If it is to be used in fermentors it should be sterilisable. This is preferably performed by autoclaving but no biosensor enzymes can presently withstand such drastic wet-heat treatment. In either case, the biosensor should not be prone to fouling or proteolysis. The complete biosensor should be cheap, small, portable and capable of being used by semi-skilled operators. There should be a market for the biosensor. There is clearly little purpose developing a biosensor if other factors (e.g. government subsidies, the continued employment of skilled analysts, or poor customer perception) encourage the use of traditional methods and discourage the decentralisation of laboratory testing. 19.2.2 Some of the notable characteristics of biosensor
Specificity: Biosensor has got the remarkable ability to distinguish between the analyte of interest and other similar substances. With biosensor, an analyte can be detected with great accuracy . Response time: Analytic tracers or catalytic product can be detected directly and instantaneously with the help of biosensors. Thus they are very much suitable for on-line monitoring. Simplicity: The sensing molecule and the transducer are integrated on to a single probe. Thus handling of biosensors is very easy. Continuous monitoring ability: Biosensors can be regenerated and reused. Thus they are suitable for continuous monitoringJ purposes which can never be done with conventional analysis methods. Reproducibility: Biosensors are very reliable; reproducibility of the values is also a great advantage. Portability: Portability of such sensors adds to the advantages which can never be the case with a spectrophotometer. Cost: The cost of the biosensor has too some extend limited its application. 19.3 Working of Biosensor Mechanism of Biosensor ________________________________________ Figure 19. 1 Schematic diagram showing the main components of a biosensor. The biocatalyst (a) converts the substrate to product. This reaction is determined by the transducer (b) which converts it to an electrical signal. The output from the transducer is amplified (c), processed (d) and displayed (e). ________________________________________ A biosensor is a device that detects, transmits and records information regarding a physiological or biochemical change. Technically, it is a probe that integrates a biological component with an electronic transducer thereby converting a biochemical signal into a quantifiable electrical response. The sensor works by converting the signal produced by the biological sensing element on response to a specific analyte to a measurable electrical signal with the help of the transducer. The amplifier increases the intensity of the signal so that it can be readily measured. The digital display then displays the reading in a suitable unit. All these components are generally integrated onto a single probe to make the handling easier. Biosensor is device that on matching with appropriate biological sample gives signal to electrical components. Biological component is bound or immobilized on the electronic component called as transducer. Figure 19. 2 The electrical signal from the transducer is often low and superimposed upon a relatively high and noisy (i.e. containing a high frequency signal component of an apparently random nature, due to electrical interference or generated within the electronic components of the transducer) baseline. The signal processing normally involves subtracting a 'reference' baseline signal, derived from a similar transducer without any biocatalytic membrane, from the sample signal, amplifying the resultant signal difference and electronically filtering (smoothing) out the unwanted signal noise. The relatively slow nature of the biosensor response considerably eases the problem of electrical noise filtration. The analogue signal produced at this stage may be output directly but is usually converted to a digital signal and passed to a microprocessor stage where the data is processed, converted to concentration units and output to a display device or data store. 19.4 Classification of Biosensor There are several type of Biosensor depends on the physical characteristic like heat , electrical signal . light, electron ,charge, and mass. Biosensors may be classified according to several criteria, such as transducers, bioactive components, or immobilization techniques used.
The heat output (or absorbed) by the reaction (calorimetric biosensors), changes in the distribution of charges causing an electrical potential to be produced (potentiometric biosensors), movement of electrons produced in a redox reaction (amperometric biosensors), light output during the reaction or a light absorbance difference between the reactants and products (optical biosensors), or Effects due to the mass of the reactants or products (piezo-electric biosensors). There are three so-called 'generations' of biosensors First generation biosensors where the normal product of the reaction diffuses to the transducer and causes the electrical response. Second generation biosensors which involve specific 'mediators' between the reaction and the transducer in order to generate improved response. Third generation biosensors where the reaction itself causes the response and no product or mediator diffusion is directly involved. A typical biosensor has got two main parts Biological component which can be enzyme, antibody, nucleic acid, microorganism, tissue, cell, etc. Electronic device, i.e., transducer which can be electrochemical, optical, piezoelectric, thermal, etc. The biological component is generally immobilized near or onto the transducer surface in order to facilitate reuse, to minimize interference and to maximize response. In each case it is the ability of the biological component to react or respond specifically to an analyte (or a group of analytes) that makes the biomolecule suitable for use as the sensing element of a biosensor. For example, an antibody will only bind to the specific antigen under suitable conditions. The biological signal that is produced is converted by means of a suitable transducer into a quantifiable electrical signal.Biosensors are suitable for detecting a wide variety of analytes including pollutants, explosives, viruses, biochemical & pharmaceutical products, vitamins, amino acids, heavy metals, ions, gases etc . In fact, every single analyte, be it simple or complex can be detected provided a biomolecule that response specifically to it is identified. 1. Transducers This is the component of biosensor which converts the biological signal to a quantifiable electrical signal. Electrochemical: In this configuration, sensing molecules are either coated onto or covalently bonded to a probe surface. A membrane holds the sensing molecule in place, excluding interfering species from the analyte solution. The sensing molecule reacts specifically with compounds to be detected, sparking an electrical signal proportional to the concentration of the analyte. The most common detection method for electrochemical biosensors involves measurement of current, voltage, capacitance, conductance and impedance. Among such sensors, amperometric (e.g. Oxygen or hydrogen peroxide) and potentiometric (e.g. pH and carbon dioxide) transducers have found the widest applications. Optical: In optical biosensors, the optical fibers allow detection of analytes on the basis of absorption, fluorescence or light scattering. Since they are nonelectrical, optical biosensors have the advantages of lending themselves to in vivo applications and allowing multiple analytes to be detected by using different monitoring wave-lengths. The versatility of fiber optics probes is due to their capacity to transmit signals that reports on changes in wavelength, wave propagation, time, intensity, distribution of the spectrum or polarity of light. Piezoelectric: In this mode, sensing molecules are attached to a piezoelectric surface-a mass to frequency transducer-in which interactions between the analyte and the sensing molecules set up mechanical vibrations that can be translated into an electrical signal proportional to the analyte. Example of such sensor is quartz crystals. Thermal: In this mode, the biocomponent is immobilized in proximity to the heat
sensing transducer, generally a thermister. Most of the enzymatic or microbial reactions are accompanied by considerable heat evolution making this sensor applicable to a very wide range of detection. However the use of sophisticated and expensive instrumentation is the major drawback of this technique. 2. Bioactive components This is the biological part of the biosensor which specifically reacts with the analyte of interest sparking a signal that is detectable by the attached transducer. Enzymes: Purified enzymes have been commonly used in the construction of biosensors due to their high specific activities as well as high analytical specificity. They are mostly used in catalytic type biosensors. Purified enzymes, however, are expensive and unstable, thus limiting their application sin the field of biosensors. As most of the enzymes being employed are intracellular, isolation and purification becomes tough. Antibodies: The binding between an antigen and its corresponding antibody is very specific. This property of antibody is exploited while designing biosensors based on antibodies. The binding reaction between the antibody and antigen can be monitored as a time dependent change of fluorescence signal which is proportional to the reaction ratio of antibody to analyte. Cells: Either whole microorganisms or tissues can be used as the biocomponent. Whole cells can be used either in a viable or non-viable form. Viable microbes metabolize various organic compounds either anaerobically or aerobically resulting in various end products like ammonia, carbon dioxide, acids etc that can be monitored using a variety of transducers. Viable cells are mainly used when the overall substrate assimilation capacity of microorganism is taken as an index of respiratory metabolic activity, as in the case of estimation of BOD, vitamins, sugars, organic acids, etc. Another mechanism used for the viable microbial biosensor involves the inhibition of microbial respiration by the analyte of interest, like environmental pollutants. The major limitation to the use of whole cells is the diffusion of substrate and products through the cell wall resulting in a slow response as compared to enzyme-based sensors. Nucleic acids: The ability of a single stranded nucleic acid to hybridize with another fragment of DNA by complementary base pairing is the principle behind the nucleic acid sensors. Technological innovation is introduced in the manner in which the nucleic acid oligomer is attached to the surface of the detector and the manner in which the hybridized nucleic acid is detected and transduced into a measurable signal. Ammonia derivetised oligonucleotides can be detected by attaching to glass surfaces such as fiber-optics cables, glass beads or microscopic slides through covalent bonding with a chemical linker. Lipids: An active biological receptor can be immobilized and stabilized in a polymeric film for determining an analyte of interest in a sample. 3. Immobilization techniques for the bio-component: The biological material should bring the physico-chemical changes in close proximity of a transducer. Immobilization not only helps in forming the required close proximity between the biomaterial and the transducer, but also helps in stabilizing it for reuse.The selection of a technique and/or support material would depend on the nature of the biomaterial and the substrate and configuration of the transducer used. Covalent binding, a commonly used technique for the immobilization of enzymes and antibodies, has not been useful for the immobilization of cells. On the other hand, cross-linking using bifunctional reagents like glutaraldehyde has been successfully used for the immobilization of cells in various supports. Thus crosslinking technique will be useful in obtaining immobilized non-viable cell preparations containing active intracellular enzymes. Entrapment and adsorption techniques are more useful when viable cells are used. The synthetic polymers used for microbial biosensor applications include polyacrylamide, polyurethane-based
hydrogels, photo cross-linkable resins and polyvinyl alcohol. Natural polymers used for the entrapment of the cells include alginate, carrageenam, low-melting agarose, chitosan, etc. But entrapment technique adds another diffusional barrier.Passive trapping of cells into the pores or adhesion onto the surfaces of cellulose or other synthetic membranes has the major advantage of direct contact between the liquid phase and the cell, thus reducing or eliminating the problem of mass transfer Table 19.1 table gives detail account of different type of Biosensor. Analyte Microorganism Transducer/immobilization Detection limit BOD Trichosporum cutaneum Miniature O2 electrode 0.2-28 mg/L BOD Activated sludge (mixed microbial consortium) O2 electrode/flow injection system >3.5 mg/L Phenolic compounds Ps. Putida O2 electrode (reactor with cells adsorbed on PEI glass) 100 M Nitrite Nitrobactor vulgaris O2 electrode (adsorption on Whatman paper) >10 M Cyanide S. cerevisiae O2 electrode (PVA) 0.15-15 nM Organophosphate nerve agent GEM E.coli Potentiometric 0.055-1.8 mM Mercuric chloride Synechococcus sp. PCC 7942 Photoelectrochemical 0.2 and 0.06 M Alcohol Candida vini O2 electrode (Porous acetyl cellulose filter) 0.20.02 mM Glucose A. niger O2 electrode (entrapment in dialysis membrane) >1.75 mM Glucose, sucrose, lactose G. oxydans, S. cerevisiae, K. marxianus O2 electrode (gelatine) Up to 0-0.8 mM Sugars (glucose) Psychophilic D.radiodurans O2 electrode (agarose) 0.03-0.55 mM Short chain fatty acids in milk A. nicotianae O2 electrode (Polyvinyl alcohol) 0.11-1.7 mM CO2 CO2 utilizing autotrophic bacteria O2 electrode (bound on cellulose nitrate membrane) 0.2-5 mM Vitamin B-6 S. uvarum O2 electrode (adsorption on cellulose nitrate membrane) 0.5-2.5 ng/ml Vitamin B-12 E. coli O2 electrode (trapped in porous acetyl cellulose membrane) 5-25x10-9 mM Peptides B. subtilis O2 electrode (filter paper strip & dialysis membrane)0.070.6 mM Phenylalanine P. vulgaris Amperometric O2 electrode 2.5x10-2 -2.5mM Pyruvate Streptococcus faecium CO2 gas sensing electrode 0.22-32 mM Tyrosine A.phenologenes NH3 gas sensing electrode 8.2x10-2 -1.0 mM Monitoring toxicity of compounds to eukaryotes S. cerevisiae was genetically modified to express firefly luciferase On-line monitoring of microbial growth E. coli engineered for constitutive bioluminescence Toxicity of Zn, Cu and Cd, alone or in combination E. coli HB101 and Ps. fluorescens 10586 genetically modified with luxCDABE Polycyclic aromatic hydrocarbons Ps. fluorescens HK44 genetically modified with luxCDABE Ecotoxicity assessment of organotins and their initial breakdown products Microtox and luxCDABE modified Ps. fluorescens Ethanol as a model toxicant E. coli TV1061, harboring the plasmid pGrpELux5 Monitoring of biocides Bioluminescent strain of E. coli produced by rDNA technology Metals, solvents, crop protection chemicals E. coli heat shock promoters, dnaK
& grpE were fused with lux gene of V. fischeri Assessment of the toxicity of metals in soils amended with sewage sludge luxCDABE modified Ps. fluorescens Ps. fluorescens 19.5Type of Biosensor 19.5.1 Optical biosensors There are two main areas of development in optical biosensors. These involve determining changes in light absorption between the reactants and products of a reaction, or measuring the light output by a luminescent process. The former usually involve the widely established, if rather low technology, use of colorimetric test strips. These are disposable single-use cellulose pads impregnated with enzyme and reagents. The most common use of this technology is for whole-blood monitoring in diabetes control. In this case, the strips include glucose oxidase, horseradish peroxidase (EC 1.11.1.7) and a chromogen (e.g. otoluidine or 3,3',5,5'-tetramethylbenzidine). The hydrogen peroxide, produced by the aerobic oxidation of glucose, oxidising the weakly coloured chromogen to a highly coloured dye. peroxidase chromogen(2H) + H2O2 dye + 2H2O [19.2] The evaluation of the dyed strips is best achieved by the use of portable reflectance meters, although direct visual comparison with a coloured chart is often used. A wide variety of test strips involving other enzymes are commercially available at the present time.A most promising biosensor involving luminescence uses firefly luciferase (Photinus-luciferin 4-monooxygenase (ATP-hydrolysing), EC 1.13.12.7) to detect the presence of bacteria in food or clinical samples. Bacteria are specifically lysed and the ATP released (roughly proportional to the number of bacteria present) reacted with D-luciferin and oxygen in a reaction which produces yellow light in high quantum yield. luciferase ATP + D-luciferin + O2 oxyluciferin + AMP + pyrophosphate + CO2 + light (562 nm) [19.3] The light produced may be detected photometrically by use of high-voltage, and expensive, photomultiplier tubes or low-voltage cheap photodiode systems. The sensitivity of the photomultiplier-containing systems is, at present, somewhat greater (< 104 cells ml-1, < 10-12 M ATP) than the simpler photon detectors which use photodiodes. Firefly luciferase is a very expensive enzyme, only obtainable from the tails of wild fireflies. Use of immobilised luciferase greatly reduces the cost of these analyses. 19.5.2 Amperometric biosensors Amperometric biosensors function by the production of a current when a potential is applied between two electrodes. They generally have response times, dynamic ranges and sensitivities similar to the potentiometric biosensors. The simplest amperometric biosensors in common usage involve the Clark oxygen electrode (Figure 19.3). This consists of a platinum cathode at which oxygen is reduced and a silver/silver chloride reference electrode. When a potential of -0.6 V, relative to the Ag/AgCl electrode is applied to the platinum cathode, a current proportional to the oxygen concentration is produced. Normally both electrodes are bathed in a solution of saturated potassium chloride and separated from the bulk solution by an oxygen-permeable plastic membrane (e.g. Teflon, polytetrafluoroethylene). The following reactions occur: Ag anode 4Ag0 + 4Cl- 4AgCl + 4e[19.4] Pt cathode O2 + 4H+ + 4e- 2H2O [19.5] The efficient reduction of oxygen at the surface of the cathode causes the oxygen concentration there to be effectively zero. The rate of this electrochemical reduction therefore depends on the rate of diffusion of the oxygen from the bulk solution, which is dependent on the concentration gradient and hence the bulk oxygen concentration. It is clear that a small, but significant, proportion of the oxygen present in the bulk is consumed by this process; the oxygen electrode
measuring the rate of a process which is far from equilibrium, whereas ionselective electrodes are used close to equilibrium conditions. This causes the oxygen electrode to be much more sensitive to changes in the temperature than potentiometric sensors. A typical application for this simple type of biosensor is the determination of glucose concentrations by the use of an immobilised glucose oxidase membrane. The reaction results in a reduction of the oxygen concentration as it diffuses through the biocatalytic membrane to the cathode, this being detected by a reduction in the current between the electrodes (Figure 19.3 ). Other oxidases may be used in a similar manner for the analysis of their substrates (e.g. alcohol oxidase, D- and L-amino acid oxidases, cholesterol oxidase, galactose oxidase, and urate oxidase) ________________________________________ Figure 19. 3. Schematic diagram of a simple amperometric biosensor. A potential is applied between the central platinum cathode and the annular silver anode. This generates a current (I) which is carried between the electrodes by means of a saturated solution of KCl. This electrode compartment is separated from the biocatalyst (here shown glucose oxidase, GOD) by a thin plastic membrane, permeable only to oxygen. The analyte solution is separated from the biocatalyst by another membrane, permeable to the substrate(s) and product(s). This biosensor is normally about 1 cm in diameter but has been scaled down to 0.25 mm diameter using a Pt wire cathode within a silver plated steel needle anode and utilising dip-coated membranes. ________________________________________ Figure 19. 4 An alternative method for determining the rate of this reaction is to measure the production of hydrogen peroxide directly by applying a potential of +0.68 V to the platinum electrode, relative to the Ag/AgCl electrode, and causing the reactions: Pt anode H2O2 O2 + 2H+ + 2e[19.6] Ag cathode 2AgCl + 2e- 2Ag0 + 2Cl-[19.7] The major problem with these biosensors is their dependence on the dissolved oxygen concentration. This may be overcome by the use of 'mediators' which transfer the electrons directly to the electrode bypassing the reduction of the oxygen co-substrate. In order to be generally applicable these mediators must possess a number of useful properties. They must react rapidly with the reduced form of the enzyme. They must be sufficiently soluble, in both the oxidised and reduced forms, to be able to rapidly diffuse between the active site of the enzyme and the electrode surface. This solubility should, however, not be so great as to cause significant loss of the mediator from the biosensor's microenvironment to the bulk of the solution. However soluble, the mediator should generally be non-toxic. The overpotential for the regeneration of the oxidised mediator, at the electrode, should be low and independent of pH. The reduced form of the mediator should not readily react with oxygen. The ferrocenes represent a commonly used family of mediators (Figure 19.5 a). Their reactions may be represented as follows, Figure 19. 5 (a) Ferrocene (5-bis-cyclopentadienyl iron), the parent compound of a number of mediators. (b) TMP+, the cationic part of conducting organic crystals. (c) TCNQ.-, the anionic part of conducting organic crystals. It is a resonancestabilised radical formed by the one-electron oxidation of TCNQH2. Electrodes have now been developed which can remove the electrons directly from the reduced enzymes, without the necessity for such mediators. They utilise a coating of electrically conducting organic salts, such as N-methylphenazinium cation (NMP+, Figure6.7b) with tetracyanoquinodimethane radical anion (TCNQ.Figure 6.7c). Many flavo-enzymes are strongly adsorbed by such organic conductors due to the formation of salt links, utilising the alternate positive and negative
charges, within their hydrophobic environment. Such enzyme electrodes can be prepared by simply dipping the electrode into a solution of the enzyme and they may remain stable for several months. Figure 19. 6 The response of an amperometric biosensor utilising glucose oxidase to the presence of glucose solutions. Between analyses the biosensor is placed in oxygenated buffer devoid of glucose. The steady rates of oxygen depletion may be used to generate standard response curves and determine unknown samples. The time required for an assay can be considerably reduced if only the initial transient (curved) part of the response need be used, via a suitable model and software. The wash-out time, which roughly equals the time the electrode spends in the sample solution, is also reduced significantly by this process. ________________________________________ These electrodes can also be used for reactions involving NAD(P)+-dependent dehydrogenases as they also allow the electrochemical oxidation of the reduced forms of these coenzymes. The three types of amperometric biosensor utilising product, mediator or organic conductors represent the three generations in biosensor development (Figure 19.6). The reduction in oxidation potential, found when mediators are used, greatly reduces the problem of interference by extraneous material. The current (i) produced by such amperometric biosensors is related to the rate of reaction (vA) by the expression: i = nFAvA (19.8) where n represents the number of electrons transferred, A is the electrode area, and F is the Faraday. Usually the rate of reaction is made diffusionally controlled by use of external membranes. Under these circumstances the electric current produced is proportional to the analyte concentration and independent both of the enzyme and electrochemical kinetics. Substrate(2H) + FAD-oxidase Product + FADH2-oxidasefi [19.9] This is followed by the processes: (a) biocatalyst FADH2-oxidase + O2 FAD-oxidase + H2O2 [19.10] electrode H2O2 O2 + 2H+ + 2e[19.11] (b) biocatalyst FADH2-oxidase + 2 Ferricinium+ FAD-oxidase + 2 Ferrocene + 2H+ [19.12] electrode 2 Ferrocene 2 Ferricinium+ + 2e[19.13] (c) biocatalyst/electrode FADH2-oxidase FAD-oxidase + 2H+ + 2e[19.14] ________________________________________
Figure 19. 7 Amperometric biosensors for flavo-oxidase enzymes illustrating the three generations in the development of a biosensor. The biocatalyst is shown schematically by the cross-hatching. (a) First generation electrode utilising the H2O2 produced by the reaction. (E0 = +0.68 V). (b) Second generation electrode utilising a mediator (ferrocene) to transfer the electrons, produced by the reaction, to the electrode. (E0 = +0.19 V). (c) Third generation electrode directly utilising the electrons produced by the reaction. (E0 = +0.10 V). All electrode potentials (E0) are relative to the Cl-/AgCl,Ag0 electrode. The following reaction occurs at the enzyme in all three biosensors: 19.5.3 Potentiometer biosensors Potentiometric biosensors make use of ion-selective electrodes in order to transduce the biological reaction into an electrical signal. In the simplest terms this consists of an immobilised enzyme membrane surrounding the probe from a pH-
meter (Figure 19.8), where the catalysed reaction generates or absorbs hydrogen ions (Table 19.2). The reaction occurring next to the thin sensing glass membrane causes a change in pH which may be read directly from the pH-meter's display. Typical of the use of such electrodes is that the electrical potential is determined at very high impedance allowing effectively zero current flow and causing no interference with the reaction. ________________________________________ Figure 19. 8 A simple potentiometric biosensor. A semi-permeable membrane (a) surrounds the biocatalyst (b) entrapped next to the active glass membrane (c) of a pH probe (d). The electrical potential (e) is generated between the internal Ag/AgCl electrode (f) bathed in dilute HCl (g) and an external reference electrode (h). ________________________________________ A. There are three types of ion-selective electrodes which are of use in biosensors Glass electrodes for cations (e.g. normal pH electrodes) in which the sensing element is a very thin hydrated glass membrane which generates a transverse electrical potential due to the concentration-dependent competition between the cations for specific binding sites. The selectivity of this membrane is determined by the composition of the glass. The sensitivity to H+ is greater than that achievable for NH4+, Glass pH electrodes coated with a gas-permeable membrane selective for CO2, NH3 or H2S. The diffusion of the gas through this membrane causes a change in pH of a sensing solution between the membrane and the electrode which is then determined. Solid-state electrodes where the glass membrane is replaced by a thin membrane of a specific ion conductor made from a mixture of silver sulphide and a silver halide. The iodide electrode is useful for the determination of I- in the peroxidase reaction (Table 19.2c) and also responds to cyanide ions. ________________________________________ Table 19.2. Reactions involving the release or absorption of ions that may be utilised by potentiometric biosensors. (a) H+ cation, glucose oxidase H2O D-glucose + O2 D-glucono-1,5-lactone + H2O2 D-gluconate + H+ penicillinase penicillin penicilloic acid + H+ [19.6] urease (pH 6.0)a H2NCONH2 + H2O + 2H+ 2NH4+ + CO2 [19.7] urease (pH 9.5)b H2NCONH2 + 2H2O 2NH3 + HCO3- + H+ [19.8] lipase neutral lipids + H2O glycerol + fatty acids + H+ [19.9] (b) NH4+ cation, L-amino acid oxidase L-amino acid + O2 + H2O keto acid + NH4+ + H2O2 [19.20] asparaginase ` L-asparagine + H2O L-aspartate + NH4+ [19.21] urease (pH 7.5) H2NCONH2 + 2H2O + H+ 2NH4++ HCO3[19.22] (c) I- anion peroxidase H2O2 + 2H+ + 2I- I2 + 2H2O [19.23] (d) CN-anion -glucosidase amygdalin + 2H2O 2glucose + benzaldehyde + H+ + CN[19.24] [19.5]
a Can also be used in NH4+ and CO2 (gas) potentiometric biosensors. b Can also be used in an NH3 (gas) potentiometric biosensor. ________________________________________ The response of an ion-selective electrode is given by (19.25) where E is the measured potential (in volts), E0 is a characteristic constant for the ion-selective/external electrode system, R is the gas constant, T is the absolute temperature (K), z is the signed ionic charge, F is the Faraday, and [i] is the concentration of the free uncomplexed ionic species (strictly, [i] should be the activity of the ion but at the concentrations normally encountered in biosensors, this is effectively equal to the concentration). This means, for example, that there is an increase in the electrical potential of 59 mv for every decade increase in the concentration of H+ at 25C. The logarithmic dependence of the potential on the ionic concentration is responsible both for the wide analytical range and the low accuracy and precision of these sensors. Their normal range of detection is 10-4 - 10-2 M, although a minority are ten-fold more sensitive. Typical response time are between one and five minutes allowing up to 30 analyses every hour. Biosensors which involve H+ release or utilisation necessitate the use of very weakly buffered solutions (i.e. < 5 mM) if a significant change in potential is to be determined. The relationship between pH change and substrate concentration is complex, including other such non-linear effects as pH-activity variation and protein buffering. However, conditions can often be found where there is a linear relationship between the apparent change in pH and the substrate concentration. A recent development from ion-selective electrodes is the production of ionselective field effect transistors (ISFETs) and their biosensor use as enzymelinked field effect transistors (ENFETs, Figure 19.9). Enzyme membranes are coated on the ion-selective gates of these electronic devices, the biosensor responding to the electrical potential change via the current output. Thus, these are potentiometric devices although they directly produce changes in the electric current. The main advantage of such devices is their extremely small size (<< 0.1 mm2) which allows cheap mass-produced fabrication using integrated circuit technology. As an example, a urea-sensitive FET (ENFET containing bound urease with a reference electrode containing bound glycine) has been shown to show only a 15% variation in response to urea (0.05 - 10.0 mg ml-1) during its active lifetime of a month. Several analytes may be determined by miniaturised biosensors containing arrays of ISFETs and ENFETs. The sensitivity of FETs, however, may be affected by the composition, ionic strength and concentrations of the solutions analysed. ________________________________________ Figure 19. 9 Schematic diagram of the section across the width of an ENFET. The actual dimensions of the active area is about 500 m long by 50 m wide by 300 m thick. The main body of the biosensor is a p-type silicon chip with two n-type silicon areas; the negative source and the positive drain. The chip is insulated by a thin layer (0.1 m thick) of silica (SiO2) which forms the gate of the FET. Above this gate is an equally thin layer of H+-sensitive material (e.g. tantalum oxide), a protective ion selective membrane, the biocatalyst and the analyte solution, which is separated from sensitive parts of the FET by an inert encapsulating polyimide photopolymer. When a potential is applied between the electrodes, a current flows through the FET dependent upon the positive potential detected at the ion-selective gate and its consequent attraction of electrons into the depletion layer. This current (I) is compared with that from a similar, but non-catalytic ISFET immersed in the same solution. (Note that the electric current is, by convention, in the opposite direction to the flow of electrons). 19.5.4 Calorimetric biosensors
Many enzyme catalysed reactions are exothermic, generating heat (Table 19.3) which may be used as a basis for measuring the rate of reaction and, hence, the analyte concentration. This represents the most generally applicable type of biosensor. The temperature changes are usually determined by means of thermistors at the entrance and exit of small packed bed columns containing immobilised enzymes within a constant temperature environment (Figure 19.10). Under such closely controlled conditions, up to 80% of the heat generated in the reaction may be registered as a temperature change in the sample stream. This may be simply calculated from the enthalpy change and the amount reacted. If a 1 mM reactant is completely converted to product in a reaction generating 100 kJ mole-1 then each ml of solution generates 0.1 J of heat. At 80% efficiency, this will cause a change in temperature of the solution amounting to approximately 0.02C. This is about the temperature change commonly encountered and necessitates a temperature resolution of 0.0001C for the biosensor to be generally useful. ________________________________________ Table 19.3. Heat output (molar enthalpies) of enzyme catalysed reactions. Reactant Enzyme Heat output -H (kJ mole-1) Cholesterol Cholesterol oxidase 53 Esters Chymotrypsin 4 - 16 Glucose Glucose oxidase 80 Hydrogen peroxide Catalase 100 Penicillin G Penicillinase 67 Peptides Trypsin 10 - 30 Starch Amylase 8 Sucrose Invertase 20 Urea Urease 61 Uric acid Uricase 49 ________________________________________ Figure 19. 10. Schematic diagram of a calorimetric biosensor. The sample stream (a) passes through the outer insulated box (b) to the heat exchanger (c) within an aluminium block (d). From there, it flows past the reference thermistor (e) and into the packed bed bioreactor (f, 1ml volume), containing the biocatalyst, where the reaction occurs. The change in temperature is determined by the thermistor (g) and the solution passed to waste (h). External electronics (l) determines the difference in the resistance, and hence temperature, between the thermistors. ________________________________________ The thermistors, used to detect the temperature change, function by changing their electrical resistance with the temperature, obeying the relationship (19.26) therefore: (19.27) Where R1 and R2 are the resistances of the thermistors at absolute temperatures T1 and T2 respectively and B is a characteristic temperature constant for the thermistor. When the temperature change is very small, as in the present case, B(1/T1) - (1/T2) is very much smaller than one and this relationship may be substantially simplified using the approximation when x<<1 that ex1 + x (x here being B(1/T1) - (1/T2), (19.28) As T1 T2, they both may be replaced in the denominator by T1. (19.29) The relative decrease in the electrical resistance (R/R) of the thermistor is proportional to the increase in temperature (T). A typical proportionality constant (-B/T12) is -4%C-1. The resistance change is converted to a proportional voltage change, using a balanced Wheatstone bridge incorporating precision wirewound resistors, before amplification. The expectation that there will be a linear correlation between the response and the enzyme activity has been found to be
borne out in practice. A major problem with this biosensor is the difficulty encountered in closely matching the characteristic temperature constants of the measurement and reference thermistors. An equal movement of only 1C in the background temperature of both thermistors commonly causes an apparent change in the relative resistances of the thermistors equivalent to 0.01C and equal to the full-scale change due to the reaction. It is clearly of great importance that such environmental temperature changes are avoided, which accounts for inclusion of the well-insulated aluminium block in the biosensor design . The sensitivity (10-4 M) and range (10-4 - 10-2 M) of thermistor biosensors are both quite low for the majority of applications although greater sensitivity is possible using the more exothermic reactions (e.g. catalase). The low sensitivity of the system can be increased substantially by increasing the heat output by the reaction. In the simplest case this can be achieved by linking together several reactions in a reaction pathway, all of which contribute to the heat output. Thus the sensitivity of the glucose analysis using glucose oxidase can be more than doubled by the co-immobilisation of catalase within the column reactor in order to disproportionate the hydrogen peroxide produced. An extreme case of this amplification is shown in the following recycle scheme for the detection of ADP. Figure 19. 11 ADP is the added analyte and excess glucose, phosphoenol pyruvate, NADH and oxygen are present to ensure maximum reaction. Four enzymes (hexokinase, pyruvate kinase, lactate dehydrogenase and lactate oxidase) are co-immobilised within the packed bed reactor. In spite of the positive enthalpy of the pyruvate kinase reaction, the overall process results in a 1000 fold increase in sensitivity, primarily due to the recycling between pyruvate and lactate. Reaction limitation due to low oxygen solubility may be overcome by replacing it with benzoquinone, which is reduced to hydroquinone by flavo-enzymes. Such reaction systems do, however, have the serious disadvantage in that they increase the probability of the occurrence of interference in the determination of the analyte of interest. Reactions involving the generation of hydrogen ions can be made more sensitive by the inclusion of a base having a high heat of protonation. For example, the heat output by the penicillinase reaction may be almost doubled by the use of Tris (tris-(hydroxymethyl)aminomethane) as the buffer.In conclusion, the main advantages of the thermistor biosensor are its general applicability and the possibility for its use on turbid or strongly coloured solutions. The most important disadvantage is the difficulty in ensuring that the temperature of the sample stream remains constant ( 0.01C). 19.5.5 Immunosensors Biosensors may be used in conjunction with enzyme-linked immunosorbent assays (ELISA). The principles behind the ELISA technique is shown in Figure 16.9. ELISA is used to detect and amplify an antigen-antibody reaction; the amount of enzymelinked antigen bound to the immobilised antibody being determined by the relative concentration of the free and conjugated antigen and quantified by the rate of enzymic reaction. Enzymes with high turnover numbers are used in order to achieve rapid response. The sensitivity of such assays may be further enhanced by utilising enzyme-catalysed reactions which give intrinsically greater response; for instance, those giving rise to highly coloured, fluorescent or bioluminescent products. Assay kits using this technique are now available for a vast range of analyses. ________________________________________ Principles of a direct competitive ELISA. (i) Antibody, specific for the antigen of interest is immobilised on the surface of a tube. A mixture of a known amount of antigen-enzyme conjugate plus unknown concentration of sample antigen is placed in the tube and allowed to equilibrate. (ii) After a suitable period the antigen and antigen-enzyme conjugate will be distributed between the bound and free states dependent upon their relative concentrations. (iii) Unbound material is washed off
and discarded. The amount of antigen-enzyme conjugate that is bound may be determined by the rate of the subsequent enzymic reaction. ________________________________________ Analyte Transducer/immobilization characteristics Low molecular weight analytes Surface plasmon resonance A monoclonal antibody against HBP is used. Lowest detection limit is 0.1 ng/ml, response time is 15 min DDT Surface plasmon resonance; covalent immobilization through an alkanethiol self-assembled monolayer (SAM) Lowest detection limit is 20 ng/L Organophosphorus Surface plasmon resonance; covalent immobilization through an alkanethiol self-assembled monolayer (SAM) Lowest detection limit is 50 ng/L Carbamate Surface plasmon resonance; covalent immobilization through an alkanethiol self-assembled monolayer (SAM) Lowest detection limit is 0.9 g/L CRP Surface plasmon resonance; Linear detection level is 2-5 g/ml using two different antiCPR antibodies Insulin with femtomole detection Liposomal immunosensors; fluorescence dye is used As low as 136 attomole. Total assay time is less than 30 min Antibiotics Poly and monoclonal antibodies are used. Recently ELISA techniques have been combined with biosensors, to form immunosensors, in order to increase their range, speed and sensitivity. A simple immunosensor configuration is shown in Figure 19.12 where the biosensor merely replaces the traditional colorimetric detection system. However more advanced immunosensors are being developed which rely on the direct detection of antigen bound to the antibody-coated surface of the biosensor. Piezoelectric and FET-based biosensors are particularly suited to such applications. ________________________________________ Figure 19. 12 Principles of immunosensors. (a)(i) A tube is coated with (immobilised) antigen. An excess of specific antibody-enzyme conjugate is placed in the tube and allowed to bind. (a)(ii) After a suitable period any unbound material is washed off. (a)(iii) The analyte antigen solution is passed into the tube, binding and releasing some of the antibody-enzyme conjugate dependent upon the antigen's concentration. The amount of antibody-enzyme conjugate released is determined by the response from the biosensor. (b)(i) A transducer is coated with (immobilised) antibody, specific for the antigen of interest. The transducer is immersed in a solution containing a mixture of a known amount of antigen-enzyme conjugate plus unknown concentration of sample antigen. (b)(ii) After a suitable period the antigen and antigen-enzyme conjugate will be distributed between the bound and free states dependent upon their relative concentrations. (b)(iii) Unbound material is washed off and discarded. The amount of antigen-enzyme conjugate bound is determined directly from the transduced signal. Piezo-electric biosensors Piezo-electric crystals (e.g. quartz) vibrate under the influence of an electric field. The frequency of this oscillation (f) depends on their thickness and cut, each crystal having a characteristic resonant frequency. This resonant frequency changes as molecules adsorb or desorb from the surface of the crystal, obeying the relationshipes (19.30) where f is the change in resonant frequency (Hz), m is the change in mass of adsorbed material (g), K is a constant for the particular crystal dependent on such factors as its density and cut, and A is the adsorbing surface area (cm2). For any piezo-electric crystal, the change in frequency is proportional to the mass of absorbed material, up to about a 2% change. This frequency change is easily detected by relatively unsophisticated electronic circuits. A simple use of such a transducer is a formaldehyde biosensor, utilising a formaldehyde dehydrogenase coating immobilised to a quartz crystal and sensitive to gaseous
formaldehyde. The major drawback of these devices is the interference from atmospheric humidity and the difficulty in using them for the determination of material in solution. They are, however, inexpensive, small and robust, and capable of giving a rapid response.
Chapter-19 Enzyme and protein STABILITY
Abstract:This report contains an overview of protein stability. The contribution each residue makes to, or takes away from, the stability of a protein is small. Thus the stability of a protein is determined by large number of small positive and negative interaction energies. In the sections that follow I discuss some of the factors that give rise to these positive and negative interaction energies. I will also briefly discuss rates of unfolding as they relate to perceived stability (Kinetic Stability),
I Definition of Stability Before entering into a discussion of stability in proteins, we must define exactly what we mean by stability. The word is used in different ways by different people. For example, a physical biochemist and a biotechnologist may each mean something different when they speak of stability. The physical biochemist, on the one hand, would probably discuss protein stability primarily in terms of the thermodynamic stability of a protein that unfolds and refolds rapidly, reversibly, cooperatively, and with a simple, two-state mechanism: Where Ku, is the equilibrium constant for unfolding. The easiest proteins in which to study folding and stability are those that exhibit this sort of rapid reversibility. Both experimental design and also theoretical treatment of data are simplified by reversible systems. Thus, it is no surprise that most of the literature reports about stability discuss this type of reversible system. The bulk of this dissertation will also focus on thermodynamic stability. In these cases, the stability of the protein is simply the difference in Gibbs free energy, G, between the folded and the unfolded states. The only factors affecting stability are the relative free energies of the folded (Gf) and the unfolded (Gu) states. The larger and more positive Gu, the more stable is the protein to denaturation. The Gibbs free energy, G, is made up the two terms enthalpy (H) and entropy (S), related by the equation:
Where T is the temperature in Kelvin. The folding free energy difference, Gu, is typically small, of the order of 5- 15 kcal/mol for a globular protein (compared to e.g. ~30 - 100 kcal/mol for a covalent bond). The biotechnologist, on the other hand, is more concerned with the practical utility of the definition: Is the protein stable enough to function under harsh conditions of temperature or solvent? While the answer to this question may lie in thermodynamic stability (discussed above); it may also lie either simply in reversibility or, for irreversibly or slowly unfolding proteins, in kinetic stability. If a protein unfolds reversibly it may be fully unfolded and inactive at high temperatures, but once it cools to room temperature, it will refold and fully recover activity. From a functional standpoint this may be all that is required for it to be classified as thermostable. However, from a thermodynamic standpoint (and in terms of this dissertation) it is classified as non-thermostable. In the case of irreversible or slowly unfolding proteins, it is kinetic stability or the rate of unfolding that is important. A protein that is kinetically stable will unfold more slowly than a kinetically unstable protein. In a kinetically stable protein, a large free energy barrier to unfolding is required and the factors affecting stability are the relative free energies of the folded (Gf) and the transition state (Gts) for the first committed step on the unfolding pathway. Kinetic stability is discussed in more detail in its own section; see Kinetic Stability. Irreversible loss of protein folded structure is represented by: Where ki is the rate constant for some irreversible inactivation process. The free energy profile for a rapidly inactivating protein is shown below. Note that once the Unfolded form is reached, the energy barrier to inactivation is lower than that to refolding.
II The Unfolded State Until recently, most studies and theoretical treatments of protein stability have assumed that the unfolded state of a protein is a random coil, an ensemble of extended conformations, in which the protein chain is extensively hydrated and the individual residues do not interact with each other. This random coil ensemble can be treated as a single state for a thermodynamic description. One consequence of this view of the unfolded state is that any mutation that effects stability must have its effect almost exclusively via the native (folded) structure. However, there is now mounting evidence that this simplistic view of the unfolded state is not always true (reviewed by Shortle, 1996). For example, small angle Xray scattering studies have shown that the radius of gyration of the unfolded state of ribonuclease A is smaller than if it is in a random coil (Sosnick & Trewhella, 1992). Further, NMR studies have shown that interactions do occur between residues, particularly between aromatic residues, in the unfolded state and that these interactions can be non-native (e.g. Pan et al, 1995). In addition, there is strong evidence that mutations can affect the degree of interaction in the unfolded state (discussed in detail in Sturtevant, 1994). For example, in one mutant of Staphylococcal nuclease, the enthalpy of unfolding, H, was reduced to 50% that of wild-type, but with no concomitant change in G for the unfolding process (Tanaka et al, 1993). (Remember that ). Although possible, to explain this result in terms of changes in the folded state alone is difficult: requiring (i) that over half the stabilizing interactions present in the wild-type be absent in the mutant and (ii) that there be a huge increase in the entropy of the (folded) mutant over the native protein. A more convincing explanation of the result is
that the enthalpy and entropy changes between mutant and wild-type are due to changes in the unfolded state, where introduction of a few interactions in the unfolded mutant could significantly decrease both enthalpy and entropy vis--vis the wild-type. There is further evidence of this type from unfolding studies with chaotropic denaturants which suggest that, for a small subset of mutants, the amount of change in accessible surface area upon unfolding is different to that in wild-type protein (Shortle & Meeker, 1986). To simplify assume that all of the intramolecular Van der Waals and hydrogen bonds that stabilize the native state are fully disrupted in the unfolded protein.
III Major Factors Affecting Protein Stability 1.hydrophobic interactions 2.hydrogen bonds 3.conformational entropy ________________________________________ The literature is in general agreement that the two types of interaction that are most prevalent in proteins are (i) hydrophobic interactions and (ii) hydrogen bonds. The reaction of these bonds upon going from the unfolded to the folded state is summarized in the cartoon below. Diagram showing the burial of hydrophobic moities and formation of intramolecular H-bonds upon protein folding. Note the release of water molecules upon folding. Although both these interactions have small free energies per residue, they are important because there are so many of them. The same is true for those interactions which stabilize the unfolded state. The most important of which is conformational entropy. Thus, the overall free energy of a folded protein is given by the small difference between two large numbers. This is a major reason for the difficulty of quantitative computational calculation of protein stability. In a recent analysis of the factors contributing to the stability of RNase T1, the stabilizing and destabilizing interactions were estimated at 271 and 286 kcal/mol, respectively (Pace et al., 1996). Hydrogen bonding and hydrophobic interactions were estimated to contribute 260 kcal/mol to the stabilizing interactions, while the bulk of the destabilizing factors were attributed to loss of conformational entropy on folding, and unfavourable burial of peptide and polar groups. See table. Destabilising Free Energy (kcal/mol) Conformational Entropy -177 Peptide Groups Buried -81 Polar Groups Buried -28 Total Destabilising -286 Stabilising Histidine Ionisation +4 Disulphide Bonds +7 Hydrophobic Groups Buried +94 Hydrogen Bonding +166 Total Stabilising +271 ----------------------------------------------------------G (estimate) -15 G (measured) +9 Note that the while the difference between the experimental and estimated values
is small, the estimated value would yield an unfolded protein. The Hydrophobic Effect The hydrophobic effect is considered to be the major driving force for the folding of globular proteins. It results in the burial of the hydrophobic residues in the core of the protein. It is exemplified by the fact that oil and water do not mix and was described well by G. S. Hartley in 1936 . "The antipathy of the paraffin chain for water is, however, frequently misunderstood. There is no question of actual repulsion between individual water molecules and paraffin chains, nor is there any very strong attraction of paraffin chains for one another. There is, however, a very strong attraction of water molecules for one another in comparison with which the paraffin-paraffin or paraffin-water attractions are slight." The thermodynamic factors which give rise to the hydrophobic effect are complex and still incompletely understood. The free energy of transfer of a non-polar compound from some reference state, such as an organic solution, into water, Gtr, is made up of an enthalpy, H, and entropy, -T S, term. At room temperature, the enthalpy of transfer from organic solution into aqueous solution is negligible; the interaction enthalpies are the same in both cases. The entropy however is negative. Water tends to form ordered cages around the nonpolar molecule and this leads to a decrease in entropy. At high temperatures (~ 110C) these cages are no longer any stronger than bulk water, and the entropy contribution tends to zero. The enthalpy of transfer, however, is now positive (unfavourable). Because the temperature dependence of entropy and enthalpy are not the same, there is some temperature at which the hydrophobic effect is strongest, and the effect decreases at temperatures above and below this temperature. The decrease in the strength of the hydrophobic effect with decreasing temperatures is probably the major cause of cold-denaturation in proteins. The contribution of the hydrophobic effect to globular protein stability has been estimated empirically both by measuring the thermodynamics of transfer of model compounds (e.g. blocked amino acids, cyclic peptides...) from organic solvents to water, and by site directed mutagenesis studies on proteins. The number arrived at is usually given as a function of the change in the solvent accessible non-polar surface area upon going from the unfolded to the folded state. The model compound studies predict that the hydrophobic effect of exposing one buried methylene group to bulk water is 0.8 kcal/mol (in Pace, 1995). The site directed mutagenesis studies yielded a larger number with greater statistical variation: the average hydrophobic effect estimated by SDM for a buried methylene group is about 1.3 kcal/mol. However, when the SDM results for methylene were plotted against the size of the cavity created by the residue substitution, and extrapolated to zero, the result at zero cavity size is 0.8 kcal/mol - in agreement with the value found for the transfer of model compounds from octanol to water (Pace et al., 1996 and references therein). In the SDM studies, cavities created by residue substitution have an additional destabilizing effect: the loss of favourable VDWs interactions (as compared to the wild-type). Thus, the "hydrophobic effect" measured by SDM includes both an entropic component due to solvent ordering and a (primarily) enthalpic component due to loss of VDWs contacts within the protein. Such an SDM study of T4 lysozyme replaced the 80% buried Ile3 residue by Val (Eriksson et al, 1992): the loss of this methyl group gave rise to a decrease in stability of 0.6 kcal/mol (corrected to 100% burial). This is smaller than expected (c.f. 0.8 kcal/mol for methylene) and suggests that the mutation introduced some smaller stabilizing influence, perhaps such as the alleviation of strain within the protein. In barnase, 15 mutants were constructed in which a hydrophobic interaction was deleted (V10A, V36A, V45A, I4A, I25A, I51A, I55A, I76A, I109A, I4V, I25V, I51V,
I55V, I76V & I109V). The finding was a strong correlation between the degree of destabilization (which ranges from 0.60 to 4.71 kcal/mol) and the number of methyl or methylene side chain groups surrounding the methyl or methylene group that was deleted (r = 0.91) (Serrano et al, 1992). See figure below. Correlation between the number of side chain methylene and methyl groups, in a radius of 6 of the group deleted from wild-type, and the changes in the free energy of unfolding for mutations of hydrophobic residues in barnase. (Taken from Serrano et al, 1992. with permission) The average free energy decrease for removal of a completely buried methylene group was found to be 1.50.6 kcal/mol. This is additive, such that Ile or Leu to Ala can destabilize a protein by up to 5 kcal/mol. (Remember that many mesophilic proteins are stable by <10 kcal/mol, so two deletions such as this would be enough to destabilize a protein completely). Hydrogen Bonds A hydrogen bond occurs when two electronegative atoms, such as nitrogen and oxygen, interact with the same hydrogen. The hydrogen is normally covalently attached to one atom, the donor, but interacts electrostatically with the other, the acceptor. This interaction is due to the dipole between the electronegative atoms and the proton. There is a geometric component involved in hydrogen bonds, and for single donor acceptor systems, such as N-H---O, the strongest hydrogen bonds are collinear (Creighton, 1993 and references therein). Electrostatic calculations suggest that deviation of 20 from linearity leads to a decrease in binding energy of approximately 10% (Pimentel & McClellan, 1960). In double acceptor systems, bifurcated hydrogen bonds with non-linear angles are preferred. The occurrence of hydrogen bonds in protein structure has been extensively reviewed by Baker & Hubbard (1984), albeit before the pdb database was as large as it is today. They found that 90% of N-H---O bonds in proteins lie between 140 and 180, and that they are centred around 158C. For C=O---H, the range is more broadly distributed between 90 and 160 and centred around 129. The strength of a hydrogen bond is between 2 and 10 kcal/mol, and one might think that this is the amount of energy one hydrogen bond contributes towards stabilization of a folded protein. However, in the unfolded state, all potential hydrogen bonding partners in the extended polypeptide chain are satisfied by hydrogen bonds to water. When the protein folds, these protein-to-water H-bonds are broken, and only some are replaced by (often sub-optimal) intra-protein Hbonds. McDonald & Thornton (1994) showed that while only 1.3% of backbone amino groups and 1.8% of carbonyl groups in proteins fail to H-bond (without any obviously compensating interactions), 80% of main chain carbonyls fail to form a second hydrogen bond. Thus, if one considers enthalpy terms alone, it would appear that hydrogen bonding is destabilizing to folded protein structure. However, one must also consider entropy. When a protein folds, and those hydrogen bonds that the protein made to bulk water are broken, the entropy of the solvent increases. The balance between the entropy and enthalpy terms are close, and in the recent past it was considered that H-bonds made no contribution overall to protein stability. But, it is now generally accepted that H-bonds make a positive contribution to protein stabilisation (reviewed in Pace et al., 1996). Estimation of the contribution of hydrogen bonding to protein stability has been made by a combination of experiments on model compounds and site-directed mutational (SDM) studies. The difficulty with the SDM studies is that when a smaller residue replaces a larger one, a cavity is created. For example, mutating Asn to Ala creates a cavity of 37.4 3 (Harpaz et al , 1994). This cavity may then be filled by water, replacing the hydrogen bonding of the asparagine NH group. Even in more conservative mutations such as Thr to Val (which is isosteric) or Ser to Ala, one must take into account the contribution of side chain entropy and the hydrophobic effect to the ( G) values. Dissection of the latter contributions
from those due only to hydrogen bonds is not trivial. An estimation has been made of a positive contribution of 1.51.0 kcal/mol (Pace et al., 1996, Fersht, 1987) from the formation of a buried intramolecular uncharged hydrogen bond. However, in order to form, the unfavourable interaction energy from burial of a polar group must be overcome. Thus, the net energy gain for formation of a buried H-bond is approximately 0.6 kcal/mol. Despite the small contribution made to protein stability by hydrogen bonds, we must remember that if we break or delete an intramolecular hydrogen bond in a protein without the possibility of forming a compensating H-bond to solvent, that protein will be destabilized. In globular proteins, much of the H-bonding potential of the backbone amide and carbonyl groups is satisfied by the formation of regular structure such as alpha helix and beta sheet (links to PPS); regular structure comprises 80 - 90% of globular protein structure. There is evidence that hydrogen bonds contribute to stability in hyperthermostable proteins. A comparison of glyceraldehyde-3-phosphate dehydrogenase (GADH) from four organisms with a range of thermostabilities and more than 50% sequence identity found that the strongest correlation to thermostability was with the number of buried charged residues H-bonded to buried neutral residues (Tanner et al, 1996). The rationalization given for this preference of charged-to-neutral over neutral-to-neutral or charged-to-charged residue H-bonding was as follows: The enthalpy of H-bond formation is in the order, charged-to-charged > charged-toneutral > neutral-to-neutral, but the entropic cost of desolvation is in the inverse order. The greatest overall free energy benefit is proposed to be for the charged-to-neutral H-bonds. Conformational Entropy of Unfolding The factor that makes the greatest contribution to stabilization of the unfolded state is its conformational entropy. It has been proposed that decreasing the conformational flexibility of the unfolded chain (by substitution with proline, or by replacement of glycine) should lead to an increase in the stability of the folded relative to the unfolded protein (Matthews et al, 1987) .Two such substitutions (A82P and G77A) in T4 lysozyme gave rise to only a small amount of stabilization (0.8 and 0.4 kcal/mol), possibly due to the counterbalancing removal of favourable interactions upon mutation.(Matthews et al, 1987) A similar mutation that also eliminated a hydrogen bond and some hydrophobic interactions was destabilizing, suggesting that these factors outweighed the decrease in conformational stability (Dixon et al, 1992). Watanabe et al (1994) have found a correlation between the number of proline residues in oligo-1,6-glucosidases from a number of bacterial species and their thermostability. Structural analysis suggests that the optimal placement is at the N-cap of alpha helices and at the second position beta type I and beta type II turns. When they substituted prolines at these positions in the homologous mesophilic enzymes they observed an increase in thermostability. However, they made their substitutions based on recruitment from the more stable enzyme. When I tried this approach in the thermostable alpha amylase from Bacillus lichinoformis, based on structural parameters alone, the mutants were either equal in stability to wildtype or destabilized (A. Day, unpublished). Structural analysis of at least one destabilized mutant (A. Day & A. Shaw unpublished) revealed the creation of a hydrophobic surface cavity upon mutation, which presumably counterbalanced any stabilizing effect of the conformational rigidity. This suggests that, in the absence of any hints from nature in terms of homology, proline substitution for stabilization should be used with some care. Watanabe et al ascribe their increase in stability to a decrease in conformational entropy of the unfolded state (c.f. the T4 lysozyme case described above by Matthews et al, 1987). As their enzyme (and ours) is irreversibly inactivated upon heating, it is equally likely that any observed increase in stability is due, not to an overall stabilization of the protein, but to a slowing of the rate of
unfolding. IV Other Factors Affecting Protein Stability There are a number of other interactions, in addition to those already discussed (The Hydrophobic Effect, Hydrogen Bonds, Conformational Entropy of Unfolding), which make contributions to the stability of proteins. These interactions are few relative to the major factors, but are still have some importance for protein stability. They include: Salt Bridges Aromatic-Aromatic Interactions Metal Binding and Disulphide Bonds Salt Bridges Salt bridges or ion-pairs are a special form of particularly strong hydrogen bonds made up of the interaction between two charged residues. The contribution of salt bridges to protein stability is a somewhat contentious issue in the literature. On the one hand is the observation that thermophilic and hyper-thermophilic analogues of mesophilic proteins tend to have increased numbers of salt-bridges (Tanner et al., 1996; Perutz & Raidt, 1975; Perutz, 1978; Dekker et al., 1991). On the other hand are mutational studies showing that the contribution of salt bridges to stability is small. Perhaps at higher temperatures salt bridges make more of a contribution to stability. Horovitz et al . (1990) measured the stability of a surface salt bridge triad between Asp8, Asp12 and Arg110 on the surface of barnase by construction of a thermodynamic cycle of all possible combinations of 1, 2, or 3 alanine mutants. The free energy contribution to the stability of the protein is only 1.25 kcal/mol for the Asp12/Arg110 pair and 0.98 kcal/mol for the Asp8/Arg110 pair. Removal of Arg 110 has no effect on stability! Other studies have had similar results (Akke & Forsen, 1990). One reason that these contributions are not as large as might be expected from the strength of such an ion pair is that, in order form a salt bridge, strong hydrogen bonds with water have to be broken. In fact, it is possible that most of the energy of stabilization comes from the increase in solvent entropy upon formation of the ion pair. In contrast to surface salt bridges, removal of one partner from a buried salt bridge leads to destabilization of 3-4 kcal/mol. However, it has been found that replacing a buried salt-bridge triad by well packed hydrophobic residues (found by random mutagenesis at the three charged residue positions) leads to an increase in stability of 4.5 kcal/mol in the arc repressor (Waldburger et al., 1995). Thus, hydrophobic interactions contribute more to stability than a salt bridge triad. This is illustrated below and is presumably due to the cost of desolvating the charged groups on going from the unfolded to the folded state. The mutant is R31M, E36Y, R40L. Interestingly, it was also shown that the wild-type arc repressor folds between 10 and 1250 more slowly than the mutant (Waldburger et al., 1996). This is proposed to be due to the high energy barrier to burying charged residues. The transition state would have a particularly high energy if one charged residue had to be buried before the other. Even if the salt bridge had formed prior to the transition state, the geometry of the salt bridge might well be sub-optimal until that part of the protein attained its native conformation. Hydrophobic interactions have less of a steric requirement. Aromatic-Aromatic Interactions About 60% of the aromatic side chains (Phe, Tyr, and Trp), found in proteins are involved in aromatic pairings. Studies with model compounds suggest that the optimal geometry is perpendicular, such that the partially positively charged
hydrogens on the edge of one ring can interact favourably with the pi electrons and partially negatively charged carbons of the other. From these observations, it might be expected that such interactions make a contribution to protein stability. This has been tested on the solvent exposed Tyr13 / Tyr17 pair on the surface of barnase (Serrano et al., 1991). Tyr13 and Tyr17 were mutated to both Ala and Phe. Mutation of the Tyr13 or Tyr17 to Phe leads to a decrease in stability of 0.30 or 0.41 kcal/mol, and 0.61 kcal/mol in the double mutant. As both hydroxyl groups are exposed to solvent (as they would be in the unfolded state), it is unclear as to the source of this small destabilization. However, mutation to the double alanine mutant leads to a decrease in stability of 4.6 kcal/mol. Most of this destabilization can be explained in terms of the loss of interactions between the tyrosine residues and the rest of the protein; however, analysis of the data using thermodynamic cycles (Carter, 1984; Horovitz & Fersht, 1990) indicates that the interaction energy between the two aromatic groups contributes only 1.3 kcal/mol to the protein stability. This is only very slightly higher than the stabilization expected from the hydrophobic contribution from burying surface area between them. In this case, therefore, there is little apparent extra stabilization due to the aromatic pair. Metal Binding Another method by which the folded state of proteins can be stabilized is metal binding, in which metal ions are coordinated, usually by lone pair donation from oxygen or nitrogen atoms. Experiments have shown that metal binding can contribute 6 - 9 kcal/mol (Braxton, 1996 and references therein) to stability. However, this is a little misleading as the comparisons made are between the apo- and the holo- enzyme; in the apo-enzyme there is often a destabilising cluster of negative charge from coordinating acidic side chains. Perhaps a fairer estimation of the contribution of metal binding to protein stability comes from an experiment in which a metal chelating site was introduced into an alpha helix of iso-cytochrome c and gave rise to a 1 kcal/mol increase in stability in the presence of saturating Cu(II) (Kellis et al., 1991). Another interesting study was that of Kuroki et al. (1989). They observed that the sequence and tertiary structure of alpha-lactalbumin (Ca2+ binding) and c-type lysozymes (non-Ca2+ binding) are homologous. They recruited the binding site from alpha-lactalbumin into human c-type lysozyme (Q86D/A92D). The mutant protein binds one mole of calcium ions and has optimal activity at about 10C higher than the wild-type. Interestingly, the apo-enzyme is about 5 C less stable than wild-type. Disulphide Bonds Disulphide bonds are formed by the oxidation of two cysteine residues to form a covalent sulphur-sulphur bond which can be intra- (examples are shown in Jane Richardson's Protein Tourist kinamage) or inter- (exemplified by insulin at this PPS link) molecular bridges. One might imagine that as the enthalpy of a covalent disulphide bond is very high, it contributes a great deal to stability. However, this bond is present in both the folded and the unfolded state, thus its enthalpic contribution to the free energy difference is negligible. All of the stabilizing effect of a disulphide bond is proposed to come from the decrease in conformational entropy of the unfolded state, as described in Conformational Entropy of Unfolding, above. Calculations suggest that a disulphide bond should give rise to 2.5 - 3.5 kcal/mol of stabilization, depending on the primary sequence separation between the crosslinks (Braxton, 1996). Experiments in which naturally occurring disulphides are either mutated to alanine, or chemically reduced and blocked, lead to decreased stability ranging from 2 - 8 kcal/mol (Betz, 1993 and references therein). Unfortunately, because these experiments also leave cavities, or buried polar groups, or otherwise have more consequences than just removal of the disulphide bridge, it is hard to estimate the stabilization due to crosslinks
alone. Introduction of novel disulphides into proteins has also had mixed results. Five disulphides have been introduced into T4 lysozyme (which has no disulphides in the wild-type). Two are destabilizing, and three are stabilizing relative to their reduced form, but all five are destabilized relative to wild-type (Betz, 1993)! However, there have been successful stabilizations by introduction of disulphides: The stability of RNase Hn is increased by 2.8 kcal/mol upon introduction of a disulphide. Engineering disulphides into the ribonuclease barnase gave rise to increased stability of 1.2 and 4.1 kcal/mol (Clarke et al., 1995). Interestingly, the former disulphide encompasses a loop of 34 residues, while the latter encompasses 17. If the stabilization is purely due to conformational entropy, the magnitude of stabilization would be the opposite way round; further, the degree of stabilization observed for the 17 residue loop (4.1 kcal/mol) is higher than would be predicted by the theory (Betz, 1993 ). Thus, it is clear that we are still far from understanding the role of disulphides in protein structure. It is worth noting that small proteins are often naturally rich in disulphide bonds, perhaps, when the geometry is optimal, they compensate for the small number of non-covalent interactions. Disulphide linkages are rare in hyper-thermostable proteins, presumably because they are chemically labile at high temperatures . Disulphides can also contribute a great deal to Kinetic Stability . V Chemical Degradation It should be mentioned that however stable a protein can be made by stabilizing the folded state, the ultimate limit of protein stability must come from covalent degradation. At high temperatures (80 - 120 C) Asn and Gln are susceptible to deamidation, Asp-Xaa peptide bonds are susceptible to hydrolysis, disulphides bonds rupture, and Xaa-Pro peptide bonds undergo cis-trans isomerisation (where Xaa is any amino acid). Interestingly, the upper limit for protein chemical thermostability may be higher than one would calculate from studies involving model mesophilic enzymes. Apart from the trivial response of just avoiding these residues (disulphides are absent and Asn/Gln content is reduced in hyper-thermophiles), it is observed that the deamidation rate of Gln and Asn residues is reduced, presumably by steric constraint, in fully folded hyper-thermophilic structures (Vielle & Zeikus, 1996 and references therein). VI Conclusions This dissertation has discussed some of the many forces that make small and conflicting contributions to protein stability. It is the sum of these various stabilising and destabilising interactions that gives rise to the final stability of a protein. The total destabilising and the total stabilising energies are both large, and their difference is small. This is one of the reasons that current computational methods struggle to predict protein stability from structure. Furthermore, our understanding of these many forces is incomplete; as often as not mutations have the opposite effect to that which had been predicted. In addition, the activation energy for folding is an important determinant of both kinetic stability and whether a protein will fold to a global minimum. However, it also clear that great strides have been made, and the search for the solution to The Folding Problem, one of the great remaining questions in biology, is and continues to be, an exciting one.
Chapter -20 Study of drug stratgies
INTRODUCTION DNA is the carrier of all genetic information in most organisms and thus a biological molecule of paramount importance. DNA replication and transcription are the basic steps in the processes of cell division and gene expression. DNA replication and transcription are regulated by several small DNA binding proteins such as transcription factors and polymerases. A number of molecules have been artificially created that mimic the interactions between the DNA and these regulatory proteins. These molecules have served as useful drugs that can be used to inibit, activate or modulate the processes of DNA replication and transcription by binding to the DNA instead of the regulatory proteins. Though the actions of activation and modulation are possible, mostly DNA is targeted in an inhibitory mode, to destroy cells for antitumor and antibiotic action. TYPES OF DNA BINDING DRUGS We are already aware of the Watson and there are two sugar phosphate backbone in an antiparallel fashion and between other. DNA binding drugs interact with NON COVALENT INTERACTIONS Crick model of DNA double helix in which strands running spirally around each other them base pairs stacked one above the DNA either non-covalently or covalently.
Non covalent interactions are generally reversible. Drugs that undergo non covalent interactions with the DNA can be classified into two main classes: Minor Groove binders Intercalators MINOR GROOVE BINDERS Minor groove binding drugs are usually shaped such that they easily fit into the groove. The binding mainly is promoted by van der Waals interactions. These drugs can also form hydrogen bonds with N-3 of adenine and O-2 of thymine. Generally minor groove binding has negligible influence on the conformation of DNA. Most minor groove binding drugs bind to A/T rich sequences. Example: Berenil. Recently, a few synthetic polyamides like lexitropsins and imidazole-pyrrole
polyamides specific forG-C and C-G regions in the grooves have been designed. These polyamides are linked systems that recognize DNA base pairs through specific orientation of imidazole (Im) and pyrrole (Py) rings. Example: The eight ring hairpin polyamide ImPyPyPy-g-ImPyPyPy-b-Dp (Dp dimethylamino propylamide) has been found to inhibit the expression of 5S RNA in fibroblast cells (skin cancer cells) by blocking the transcription factor IIIAbinding site. INTERCALATORS Intercalating agents contain planar heterocyclic groups which plug in between adjacent DNA base pairs. The complex, thus formed, is mainly stabilized by - stacking interactions between the drug and DNA bases. Triostin A has two quinoxaline units connected by a cyclic peptide structure with a cystein pair (disulfide bond) at its center. Chemical structure of triostatin A
Triostin A is an antibiotic possesing cross-linked octapeptide rings attached to two quinoxaline chromophores. The chromophores spaced at 3.5A, sandwich two base pairs between the two quinoxalines. Intercalators induce strong structural distortions in DNA. For example, Daunomycin is a DNA intercalator that is site specific for GC base pairs. On intercalation of DNA by Daunomycin the following structural changes occur: CG and GC base pairs bend by ca. 9o and 15o respectively to prevent excessive van der Waals contacts. Also, the base pairs are wedged out to a separation of 6.8 . These distortions lead to a total DNA unwinding angle of ca. 8o. These changes have the effect of inhibiting the association of DNA with the DNA helicase and topoisomerase.
COVALENT INTERACTIONS Covalent binders bind the DNA irreversibly, invariably leading to complete inhibition of DNA processes and subsequent cell death Important drugs belonging to this category are: Mitomycin C - antitumor antibiotic The activated antibiotic forms a cross links guanine bases on adjacent strands of DNA thereby inhibiting single strand formation. Anthramycin - antitumor antibiotic It binds covalently to N-2 of guanine located in the minor groove of DNA. Cisplatin (cis-diamine-dichloro-platinum) - anticancer drug. It platinates N-7 of guanine on the major groove site of DNA double helix. This leads to the cross-linking of two adjacent guanines on the same DNA strand hindering the mobility of DNA polymerases. FACTORS AFFECTING DRUG-DNA INTERACTIONS
BASE PAIR SEQUENCE Base pair sequence plays a large role on the specific nature of most DNA binding drugs. The nature and strength of the interaction occurring between the drug and the DNA depends greatly upon the the specific bases involved. The predisposition of minor groove binders like Berenil towards A/T rich regions could be explained by the following facts: A/T regions are narrower than G/C regions, thus, better van der Waals contacts. High steric hindrance in the G/C groove regions, due to the C-2 amino group of the guanine base. In the case of intercalator triostin A it has been proposed that the linker peptide structure could be responsible for specific interaction with the DNA surface. The major group specific readout sequence of H-bond donor and acceptor could be involved in triostatin A binding. COUNTER IONS DNA carries a large negative charge because of the phosphate backbone. Therefore, small positively charged counter ions such as Na+, or Ca++ and Mg++ ions may be present around the DNA. The counter ions can effectively screen the negative backbone surface thereby allowing non electrolytes as well as positively charged drug ligands to interact more strongly with the target base pairs. SOLVENT In the process of drug DNA binding, a displacement of solvent from the binding site on both the DNA and drug takes place. There also occurs a partial compensation of charges as the DNA and drug are oppositely charged which causes some partially solvated counterions to be released into the bulk solvent and become fully solvated. Hydrophobic effect is the major driving force of hydrophobic ligand receptor interaction. It is seen in the case of intercalating drugs as the hydrophobic, aromatic sidechains interact favorably with the aromatic environment of the base pair stacking. STRUCTURAL MODIFICATIONS So that the DNA and the drug molecule can accommodate each other, some structural deformation/adaptation occurs in both. Thus, it can be concluded that some of the important forces contributing to drug DNA interactions are- hydrophobic force, hydration/dehydration energies, van der waals interactions and hydrogen bonds, ion effects, etc.
CHEMOTHERAPY- DNA BINDING DRUGS AGAINST CANCER Cancer cells exhibit rapid cell division. They lose the property of contact inhibition and do not undergo appoptosis. Chemotherapy makes use of DNA binding drugs to kill cancerous cells. The chemotherepeutic drugs bind to the DNA of the cancer cells and halt the cell division. When the cancer cells fail to divide they die, ultimately causing the entire tumor to shrink. Some of the important DNA
binding drugs used in chemotherapy are cisplatin, amifosatine and mitomycin C. SIDE EFFECTS OF CHEMOTHERAPY Current mechanisms target chemotherapeutic drugs to cells that are rapidly divivding. Unfortunately, chemotherapy cannot differentiate cancer cells from healthy cells. As a result, fast dividing healthy cells such as stem cells also become severely affected by the drugs. Side effects caused by chemotherapt may be from mild to severe such as: Hair loss Nausea Low RBC count Decreased immunity Faliure of certain organs Even death. COUNTERACTING SIDE EFFECTS DESIGNING MORE ACCURATE DRUG DELIVERY STRATEGIES* For designing better drug delivery strategies, we should focus upon the differences between cancer cells and normal cells at olecular level. Conventional methods target rapidly dividing cells. The disadvantge of this approach is that, it cannot distinguish cancer cells from the stem cells, which leads to a number serious side effects. The properties of cancer cells that are unique to them should be exploited for drug targetting. A few moleculare features of cancer cells that may serve as molecular targets in chemotherapy are listed belowCancer cells have been found to be much less adhesive to other cells and non cellular substrates. It is this lack of adhesiveness that permits cancer cells to metastasize. This happens because of the expression of new cell surface proteins reffered to as tumor associated antigens in cancer cells that can induce modiications at the cell surface, thereby leading to the loss of adhesiveness. Cancer cells continue to divide indefinitely, without undergoing ageing this seemingly immortal nature of cancer cells can be attributed to the presence of the telomerase enzyme which is absent in normal cells. The telomerase enzyme matains teloeres at the ends of the chromosomes, thus allowing cells to continue to divide. Cancer cells require large amounts of glucose for energy production and growth. When cells become cancerous, they require more energy and the level of proteins needed for glucose transport and metabolism increases. As a tumor grows, it rapidly outgrows its blood supply, leaving portions of the tumor with regions where the oxygen concentration is significantly lower than in healthy tissues. As a consequence, tumor cells in these hypoxic zones rely on glycolysis for energy production and therefore further increase the levels of proteins responsible for glucose transport and metabolism. These differences could be employed for transpoting the DNA binding drugs to the cancer cells without affecting the healthy cells of the body. DESTABLISING DRUG DNA INTERACTIONS IN HEALTHY STEM CELLS*
A DNA binding drug binds with the DNA based on simple rules of chemistry. The interactions that stablise most drug-DNA complexes are simple hydogen bonds, hydrophobic effects and electrostatic interations. The whole concept rounds upto the fact that the product (drug DNA complex) must be more stable than the starting elemnt - unbound drug. If we could design synthetic molecules that offered a particular drug a set of more stablising interactions inside the cell then it could be thought of using this molecule to destablise the drug-DNA interactions in the healthy cells so that they can resume their normal cell cycle. Let us consider a situation for understanding this conept better. Let us assume that a patient of cancer was administerred with anthracycline(an inercalator), which due to inefficient drug delivery found its way into stem cells and got bound to their DNA. Now, side effects begin to manifest. In order to nullify the side effects a synthetic molecule is designed that provides a greater affinity to the drug bound to stem cell DNA than the DNA itself. This synthetic agent can now be targetted to the stem cells where it will act so as to pull anthracycline from the DNA and itself form a stabler complex with it. Now the stem cells are free of the drug and can undergo their normal cellular functions.
CONCLUSION Thus, we see that DNA binding drugs of all categories have displayed a promising potential in the treatment various deadly viral diseases and cancers. For this reason they have become targets of clinical research across the globe. DNA binding drugs such as cis-Platin have revolutionised chemotherapy so that now it is possible to completely root out cancers. However, there several hurdles still to be cleared before these drugs can be considered as a safe cure for cancer. The scientific community understands this and therefore full fledged research work is being conducted in several big research institutes and laborataries to design better and safer drugs.
SUMMARY
Certain molecules are able to mimic the naturally occuring DNA binding proteins such as transcription factors and DNA polymerases. These molecules are used as drugs that can be bound physically to the DNA for interrupting key cellular activities such as DNA replication and gene expression. DNA drug interactions can be covalent or non covalent. DNA binding drugs can be grouped into 3 categories minor groove binders, intercalators and covalent binders. Important forces contributing to drug DNA interactions are- hydrophobic force, hydration/dehydration energies, van der waals interactions and hydrogen
bonds, ion effects, etc Sveral DNA binding drugs have played an invaluable role in cancer chemotherapy, such as cis-Platin, daunomycin, and so on. Side effects of chemotherapy can be minimised by designing strategies for efficient drug targetting and destabilising the drug-DNA complexes in healthy cells. Assignment Structural Biology DNA And Drug Interaction
Introduction to DNA Deoxyribonucleic acid, DNA, is a molecule of great biological significance. The total DNA content of a cell is termed the Genome. The Genome is unique to an organism, and is the information bank governing all life processes of the organism, DNA being the form in which this information is stored. Stretches of DNA called genes have the extremely important function of coding for proteins. The function of the rest of the genome, loosely termed as non-gene regions, is not very clearly known. Fig.1 The DNA molecule DNA has two main functions, 1. Transcription: Information is retrieved from the DNA by ribonucleic acid, RNA, and utilized to synthesize proteins in the body. Proteins are involved in all body processes and play many roles. e.g. as hormones, enzymes, carriers, structural proteins, receptors, regulators etc.
2. Replication: DNA is responsible for its own regeneration, i.e., DNA self replicates. DNA is present in the body in the form of a double helix, where each strand is composed of a combination of four nucleotides, adenine (A), thymine (T), guanine (G) and cytosine (C). Within a strand these nucleotides are connected via phosphodiester linkages. The two strands are held together primarily via Watson Crick hydrogen bonds where A forms two hydrogen bonds with T and C forms three hydrogen bonds with G (Figure2). base pairing fig.2 Watson Crick Base pairing, A-T and G-C
Specific recognition of DNA sequences by proteins/ small molecules is achieved via the combination of hydrogen bond acceptor/donor sites available on the major groove or minor groove. e.g. the A-T base pair offers a hydrogen bond acceptor, N7, a donor N6, and an acceptor, O4 on the major groove side.
Transcription and replication are vital to cell survival and proliferation as well as for smooth functioning of all body processes. DNA starts transcribing or replicating only when it receives a signal, which is often in the form of a regulatory protein binding to a particular region of the DNA. Thus, if the binding specificity and strength of this regulatory protein can be mimicked by a small molecule, then DNA function can be artificially modulated, inhibited or activated by binding this molecule instead of the protein. Thus, this synthetic/natural small molecule can act as a drug when activation or inhibition of DNA function is required to cure or control a disease (Table 1). DNA and Drug Binding DNA activation would produce more quantities of the required protein, or could induce DNA replication; depending on which site the drug is targeted. DNA inhibition would restrict protein synthesis, or replication, and could induce cell death. Though both these actions are possible, mostly DNA is targeted in an inhibitory mode, to destroy cells for antitumor and antibiotic action. Drugs bind to DNA both covalently as well as non-covalently. Covalent Binding Drugs Covalent binding in DNA is irreversible and invariably leads to complete inhibition of DNA processes and subsequent cell death. Cis-platin (cisdiamminedichloroplatinum) is a famous covalent binder used as an anticancer drug, and makes an intra/interstrand cross-link via the chloro groups with the nitrogens on the DNA bases. Cisplatin, cisplatinum or cis-diamminedichloroplatinum(II) (CDDP) is a platinumbased chemotherapy drug used to treat various types of cancers, including sarcomas, some carcinomas (e.g. small cell lung cancer, and ovarian cancer), lymphomas and germ cell tumors. It was the first member of its class, which now also includes carboplatin and oxaliplatin.
Upon administration, a chloride ligand undergoes slow displacement with water (an aqua ligand) molecules, in a process termed aquation. The aqua ligand in the resulting [PtCl(H2O)(NH3)2]+ is easily displaced, allowing cisplatin to coordinate a basic site in DNA. Subsequently, the platinum cross-links two bases via displacement of the other chloride ligand.[1] Cisplatin crosslinks DNA in several different ways, interfering with cell division by mitosis. The damaged DNA elicits DNA repair mechanisms, which in turn activate apoptosis when repair proves impossible. Non-covalently bound drugs : 1. Minor groove binders- Minor groove binding drugs are usually crescent shaped, which complements the shape of the groove and facilitates binding by promoting van der Waals interactions. Additionally, these drugs can form hydrogen bonds to bases, typically to N3 of adenine and O2 of thymine. Most minor groove binding drugs bind to A/T rich sequences.
This preference in addition to the designed propensity for the electronegative pockets of AT sequences is probably due to better van der Waals contacts between the ligand and groove walls in this region, since A/T regions are narrower than G/C groove regions and also because of the steric hindrance in the latter, presented by the C2 amino group of the guanine base. However, a few synthetic polyamides like lexitropsins and imidazole-pyrrole polyamides have been designed which have specificity for G-C and C-G regions in the grooves. 2. Intercalators- These contain planar heterocyclic groups which stack between
adjacent DNA base pairs. The complex, among other factors, is thought to be stabilized by - stacking interactions between the drug and DNA bases. Intercalators introduce strong structural perturbations in DNA.
Non-covalent binding is reversible and is typically preferred over covalent adduct formation keeping the drug metabolism and toxic side effects in mind. However, the high binding strength of covalent binders is a major advantage. Proteins are large molecules and bind quite strongly to the DNA, with binding constants in the nanomolar range. It has been difficult to achieve similar specificity and affinity using small non-covalent binders, and remains a major challenge to the design of drugs for DNA. Some DNA binders are listed in the following table Table 1. Drug, action and mode of binding for some DNA binding drugs. SNo 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Drug Action Mode of Binding PDB Hoechst 33258 Antitumor Minor groove binding 264D Netropsin Antitumor, Antiviral Minor groove binding 121D Pentamidine Active against P. carinii Minor groove binding 1D64 Berenil Antitrypanosomal Minor groove binding 1D63 Guanyl bisfuramidine Active against P. carinii Minor groove binding 227D Netropsin Antitumor, Antiviral Minor groove binding 121D Distamycin Antitumor, Antiviral Minor groove binding 2DND SN7167 Antitumor, Antiviral Minor groove binding 328D SN6999 Active against P. falciparum Minor groove binding 144D Nogalamycin Antitumor Intercalation 182D Menogaril Antitumor- Topoisomerase II poison Intercalation 202D Mithramycin Anticancer antibiotic Minor groove binding 146D Plicamycin Anticancer antibiotic Minor groove binding 1BP8 Chromomycin A3 Anticancer antibiotic Minor groove binding 1EKH cis -Platin Anticancer antibiotic Covalent cross-linking 1AU5
Forces involved in DNA-drug recognition: Understanding the forces involved in the binding of proteins or small molecules to DNA is of prime importance due to two major reasons. Firstly, the design of sequence specific drugs having requisite affinity for DNA requires a knowledge how the structure of the drug is related to the specificity/affinity of binding and what structural modifications could result in a drug with desired qualities. Secondly, identifying the forces/energetics involved in such processes is fundamental to unraveling the mystery of molecular recognition in general and DNA binding in particular. Some of the forces that are known to contribute to biomolecular recognition and also to DNA-drug binding are direct electrostatic interactions, direct van der Waals/packing interactions, complex hydration/dehydration contributions composed of hydrophobic component, solvation electrostatics, solvation van der Waals, ion effects and entropy terms. DNA-drug binding may be described in the following manner,
Consider DNA-drug binding in an aqueous environment. DNA is polyanionic in nature and the drug molecule is also often charged. The associated counterions lie near the charged groups and are also partially solvated. When binding occurs, it results in a displacement of solvent from the binding site on both the DNA and drug. Also, since there would be partial compensation of charges as the DNA and drug are oppositely charged, some counterions would be released into the bulk solvent and are solvated fully. Also, the binding process would be associated with some structural deformation/adaptation of the DNA as well as the drug molecule in order to accommodate each other. All these events are associated with some energetic gains/losses, the comprehensive estimation of which is a major challenge. We are attempting to understand the energetics of DNA-drug interaction by theoretically estimating the above contributions employing classical and statistical mechanical methods. Developing a theoretical protocol for detailed quantitative analysis of DNA-ligand binding in solution is a daunting task due to some major challenges. Simulations of DNA with solvent and the attendant counterion atmosphere require careful consideration to ensure system stability. Also, evolving a computationally efficient technique using statistical mechanical principles for quantitative estimates of binding free energies in large biomolecular systems is an equally challenging task. Our study is aimed at providing such a theoretical protocol for complementing experimental techniques and facilitating a minute study of the structure-energy relationships in DNA-drug complexes. Structural and conformational changes in the DNA and drug on binding in solution are associated with enthalpic and entropic contributions to the binding free energy, which can be theoretically estimated from ensembles of structures generated via simulations. The only drawback of this approach is the long time taken for the simulations. The other terms, namely, electrostatics, van der Waals, hydrophobic component, rotational and translational entropy can be estimated from single structures. The web tool, PreDDICTA, estimates the components of DNA-drug binding free energy which can be calculated from a single structure, and correlates it with experimental binding free energy and Tm, thus providing a swift method for evaluation of potential lead candidates for researchers pursuing structure based drug design for DNA.
References and Further Reading 1. Neidle, S., Thurston, D.E. (2005) Chemical approaches to the discovery and development of cancer therapies Nat Rev Cancer, 5, 285-96. 2. Geierstanger, B.H., Wemmer, D.E. (1995) Complexes of the minor groove of DNA. Annu. Rev. Biophys. Biomol. Struct., 24, 463-493. 3. Chaires, J. B. (1998) Drug--DNA interactions. Curr. Opin. Struc. Biol., 8, 314320. 4. Neidle, S. (2001) DNA minor-groove recognition by small molecules. Nat. Prod. Rep., 18, 291-309. 5. Hurley, L. H. (2002) DNA and its associated processes as targets for cancer therapy. Nature Reviews Cancer, 2, 188-200. 6. Wemmer, D. E., Dervan, P. B. (1997) Targeting the minor groove of DNA. Curr.
Opin. Struc. Biol., 7, 355-61. 7. Jones, S, van Heyningen, P, Berman, H. M., Thornton, J. M. (1999) Protein-DNA interactions: A structural analysis. J. Mol. Biol., 287, 877-896. 8. Jen-Jacobson, L. (1997) Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers, 44,153-180. 9. Janin, J. (1999) Wet and dry interfaces: the role of solvent in protein-protein and protein-DNA recognition. Structure Fold Des., 7, R277-279. 10. Turner, P. R., Denny, W. A. (2000) The genome as a drug target: sequence specific minor groove binding ligands Curr. Drug Targ., 1, 1-14. 11. Goodsell, D. S. (2001) Sequence recognition of DNA by lexitropsins. Curr. Med. Chem., 8, 509-516. 12. Reddy, B. S., Sondhi, S. M., Lown, J. W. (1999) Synthetic DNA minor groovebinding drugs. Pharmacol. Ther., 84, 1-111. 13. Dervan, P. B., Edelson, B. S. (2003) Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr. Opin. Struct. Biol., 13, 284-299. 14. Jayaram, B., Beveridge, D.L. (1996) Modeling DNA in aqueous solutions: theoretical and computer simulation studies on the ion atmosphere of DNA. Annu. Rev. Biophys. Biomol. Struct., 25, 367-394. 15. Auffinger, P., Westhof, E. (1998) Simulations of the molecular dynamics of nucleic acids. Curr. Opin. Struct. Biol., 8, 227236. STRUCTURAL BIOLOGY ASSIGNMENT DNA-DRUG INTERACTION
MADE BY: Name: VASU R SAH No.: 38038 Semester: Vth (AIB) DNA Drug Interaction Deoxyribonucleic acid, DNA, is a molecule of great biological significance. The total DNA content of a cell is termed the Genome. The Genome is unique to an organism, and is the information bank governing all life processes of the organism, DNA being the form in which this information is stored. Stretches of DNA Roll Section: R
called genes have the extremely important function of coding for proteins. The function of the rest of the genome, loosely termed as non-gene regions, is not very clearly known. Fig.1 the DNA molecule DNA has two main functions: 1. Transcription: Information is retrieved from the DNA by ribonucleic acid, RNA, and utilized to synthesize proteins in the body. Proteins are involved in all body processes and play many roles. e.g. as hormones, enzymes, carriers, structural proteins, receptors, regulators etc. 2. Replication: DNA is responsible for its own regeneration, i.e., DNA self replicates. DNA as carrier of genetic information is a major target for drug interaction because of the ability to interfere with transcription (gene expression and protein synthesis) and DNA replication, a major step in cell growth and division. The latter is central for tumorigenesis and pathogenesis. There are three principally different ways of drug binding: First, through control of transcription factors and polymerases. Here, the drugs interact with the proteins that bind to DNA. Second, through RNA binding to DNA double helices to form nucleic acid triple helical structures or RNA hybridization (sequence specific binding) to exposed DNA single strand regions forming DNA-RNA hybrids that may interfere with transcriptional activity. Third, small aromatic ligand molecules that bind to DNA double helical structures by (i) intercalating between stacked base pairs thereby distorting the DNA backbone conformation and interfering with DNA-protein interaction or (ii) the minor groove binders. The latter cause little distortion of the DNA backbone. Both work through non-covalent interaction. The small ligand drug approach offers a simple solution. The synthesis and screening of synthetic compounds that do not exist in nature, work much like pharmacological ligand for cell surface receptors in excitable tissue, and appear to be more readily delivered to cellular targets than large RNA or protein ligands. The lack of sequence specificity for intercalating molecules, however, does not allow to target specific genes, but rather certain cellular states or physiological and pathological conditions, like rapid cell growth and division that can be selectively suppressed as compared to non growing or slowly growing healthy tissue. DNA is present in the body in the form of a double helix, where each strand is composed of a combination of four nucleotides, adenine (A), thymine (T), guanine (G) and cytosine (C). Within a strand these nucleotides are connected via phosphodiester linkages. The two strands are held together primarily via Watson Crick hydrogen bonds where A forms two hydrogen bonds with T and C forms three hydrogen bonds with G (Figure2). Fig.2 Watson Crick Base pairing, A-T and G-C base pairing Specific recognition of DNA sequences by proteins/ small molecules is achieved via the combination of hydrogen bond acceptor/donor sites available on the major groove or minor groove. e.g. the A-T base pair offers a hydrogen bond acceptor, N7, a donor N6, and an acceptor, O4 on the major groove side. DNA-Drug Interaction :
Transcription and replication are vital to cell survival and proliferation as well as for smooth functioning of all body processes. DNA starts transcribing or replicating only when it receives a signal, which is often in the form of a regulatory protein binding to a particular region of the DNA. Thus, if the binding specificity and strength of this regulatory protein can be mimicked by a small molecule, then DNA function can be artificially modulated, inhibited or activated by binding this molecule instead of the protein. Thus, this synthetic/natural small molecule can act as a drug when activation or inhibition of DNA function is required to cure or control a disease (Table 1). DNA activation would produce more quantities of the required protein, or could induce DNA replication; depending on which site the drug is targeted. DNA inhibition would restrict protein synthesis, or replication, and could induce cell death. Though both these actions are possible, mostly DNA is targeted in an inhibitory mode, to destroy cells for antitumor and antibiotic action. Drugs bind to DNA both covalently as well as non-covalently. Covalent binding in DNA is irreversible and invariably leads to complete inhibition of DNA processes and subsequent cell death. Cis-platin (cisdiamminedichloroplatinum) is a famous covalent binder used as an anticancer drug, and makes an intra/interstrand cross-link via the chloro groups with the nitrogens on the DNA bases.
Non-covalently bound drugs mostly fall under the following two classes: 1. Minor groove bindersAdditionally, these drugs can form hydrogen bonds to bases, typically to N3 of adenine and O2 of thymine. Most minor groove binding drugs bind to A/T rich sequences. This preference in addition to the designed propensity for the electronegative pockets of AT sequences is probably due to better van der Waals contacts between the ligand and groove walls in this region, since A/T regions are narrower than G/C groove regions and also because of the steric hindrance in the latter, presented by the C2 amino group of the guanine base. However, a few synthetic polyamides like lexitropsins and imidazole-pyrrole polyamides have been designed which have specificity for G-C and C-G regions in the grooves. 2. IntercalatorsThese contain planar heterocyclic groups which stack between adjacent DNA base pairs. The complex, among other factors, is thought to be stabilized by - stacking interactions between the drug and DNA bases. Intercalators introduce strong structural perturbations in DNA.
Non-covalent binding is reversible and is typically preferred over covalent adduct formation keeping the drug metabolism and toxic side effects in mind. However, the high binding strength of covalent binders is a major advantage. Proteins are large molecules and bind quite strongly to the DNA, with binding constants in the nanomolar range. It has been difficult to achieve similar specificity and affinity using small non-covalent binders, and remains a major challenge to the design of drugs for DNA.
Modeling DNA-ligand interaction of intercalating ligands: The following properties have been identified as important for the successful modeling of ligand-DNA interaction: Degrees of freedom Role of base pair sequence Counter ion effects Role of solvent ligand-receptor binding Degrees of freedom this problem is analogous to that of protein ligand interaction. The major requirement for intercalating agents is the planar aromatic ring structure. This structure fits between to adjacent base pair planes and can have some, although much restricted, rotational freedom within the plane of the ring. The ligand itself may have flexibility of structural parts outside the DNA binding site and may contain more than one intercalating sidechain: The structure of the antibiotic triostin A shows the presence of two quinoxaline (groups to the right; double aromatic rings) units linked through a cyclic peptide structure (center left) which is stabilized at its center by a cystein pair (disulfhydril covalent bond).
Fig. Chemical structure of triostatin A The space filled side view indicates how the two quinoxaline rings are positioned by the linker peptide in co-planar fashion suitable for intercalating with DNA base pairs. As a rule, the more intercalating sidechains are linked within a single ligand structure, the stronger the expected binding affinity. Triostatin A belongs to a family of antibiotics which are characterized by crosslinked octapeptide rings bearing two quinoxaline chromophores. Since the spacing between the chromophores is 3.5A, the intercalation process sandwiches two base pairs between the two quinoxalines. This phenomenon is called bis-intercalation and has first been described for echinomycin by showing that bis-intercalating drugs cause twice the DNA helix extension and unwinding seen as compared to single intercalating molecule like ethidium. The latter is a chromophore which is activated by UV light and is used by molecule biologists to label nucleic acids in gel electrophoresis or ion gradient centrifugation. Role of base pair sequence Experimental evidence suggests that base pair sequence does not play a large role on the specific mature of most intercalating complexes. As the structure of triostatin A suggests, however, the linker peptide structure may well promote specific interaction with the DNA surface. The major group specific readout sequence of H-bond donor and acceptor could be involved in triostatin A binding. The figure graphically shows the direct readout of the DNA base sequence on a double helical structure. Fig. Overlap of AT and GC base pairs The following characteristics of non covalent bond formation are associated with the binding sites indicated above: binding site GC base pair AT base pair W1 H-bond acceptor H-bond acceptor W2 blank blank
W3 H-bond acceptor H-bond donor W3' blank blank W2' H-bond donor H-bond acceptor W1' C-H weak hydrophobic CH3, strong hydrophobic While the interaction on the major groove side is distinct for the direction of the base pair (e.g. AT vs TA), there is no directionality at the minor groove side. The molecular basis of specific recognition between echinomycin and DNA is due to the hydrogen bonding between the ligand alanine carbonyl groups and the 2-amino group of guanine. This is consistent with the observation that the preferred binding site is the sequence CG. Counter ion effect DNA is a negatively charged polyanion attracting counter ions, positively charged Na+, or Ca++ and Mg++ ions as well as basic residues of proteins. The presence of small counter ion affect drug binding, since the counter ions can screen and shield the negative backbone surface allowing non electrolytes as well as positively charged ligand to interact more strongly with the DNA target. High ionic strength, however, reduces non covalent interaction mediated by hydrogen bonds and electrostatic interactions. Role of solvent ligand-receptor binding There are three general classes of interactions that must be considered in solvated ligand-receptor binding : (a) ligand solvent interaction (e.g. hydration shell), (b) receptor solvent interaction, and (c) ligand-DNA complex with solvent interaction. The three classes basically describe the sequence of events of free ligand interacting with its receptor and the change in overall solvent interaction before and after binding. We have seen that the hydrophobic effect is completely described by this system and the contribution of the entropy of free bulk water is the major driving force of hydrophobic ligand receptor interaction. This type of interaction is found in intercalating substrates because the hydrophobic, aromatic sidechains interactive favorably with the aromatic environment of the base pair stacking. The total amount of surface bound water is reduced in the after complex formation. Rational for drug design When a compound intercalates into nucleic acids, there are changes which occur in both the DNA and the compound during complex formation that can be used to study the ligand DNA interaction. The binding is of course an equilibrium process because no covalent bond formation is involved. The binding constant can be determined by measuring the free and DNA bound form of the ligand. Since many of the intercalating substrates are aromatic chromophores, this can be done spectroscopically. Also, DNA double helix structures are found to be more stable with intercalating agents present and show a reduced heat denaturation. Correlating these biophysical parameters with cytotoxicity is used to support the antitumor activity of these drugs as based on their ability to intercalate in DNA double helical structures. Improvement of anticancer drugs based on intercalating activity is not only focussed on DNA-ligand interaction, but also on tissue distribution and toxic side effects on the heart (cardiac toxicity) due to redox reduction of the aromatic rings and subsequent free radical formation. Free radical species are thought to induce destructive cellular events such as enzyme inactivation, DNA strand cleavage and membrane lipid peroxidation. Modeling DNA-ligand interaction of minor groove binders: Hairpin minor grove binding molecules have been identified and synthesized that bind to GC reach nucleotide sequences. Hairpin polyamides are linked systems that exploit a set of simple recognition rules for DNA base pairs through specific orientation of imidazole (Im) and pyrrole (Py) rings. The hairpin polyamides originated from the discovery of the three-ring Im-Py-Py molecule that bound to
minor groove DNA as an antiparallel side by side dimer. Fig. Structure of hairpin ligand (right) on DNA minor groove (left)
The compound was found to recognize GC base pairs. Solid phase synthesis of polyamides of variable length has produced efficient ligands, e.g. the eight ring hairpin polyamide ImPyPyPy-g-ImPyPyPy-b-Dp (Dp dimethylamino propylamide) shown in the figure above. This small synthetic molecule has an binding constant in the order of 0.03nM. The optimal goal of polyamide ligand design has been reached with finding structures able to recognize DNA sequences of specific genes. The structure shown above inhibits the expression of 5S RNA in fibroblast cells (skin cancer cells) by interfering with the transcription factor IIIA-binding site. A new strategy of rational drug design exploits the combination of polyamides with bis-intercalating structures. WP631 is a dimeric analog of the clinically proven anthracycline antibiotic daunorobuicin. Fig. Structure of WP631 This new synthetic compound shows an affinity of 10pM and also showed to be resistant against multidrug resistance mechanisms often encountered in antitumor therapy. Multidrug resistance is a phenomenon where small aromatic compounds are efficiently expelled from the cell by cell membrane transport proteins commonly referred to as ABC transporters (or ATP Binding Cassette proteins). Drugs that form covalent bonds with DNA targets Drugs that interfere with DNA function by chemically modifying specific nucleotides are Mitomycin C, Cisplatin, and Anthramycin. Mitomycin C is a well characterized antitumor antibiotic which forms a covalent interaction with DNA after reductive activation. The activated antibiotic forms a cross-linking structure between guanine bases on adjacent strands of DNA thereby inhibiting single strand formation (this is essential for mRNA transcription and DNA replication). Anthramycin is an antitumor antibiotic which bind covalently to N-2 of guanine located in the minor groove of DNA. Anthramycin has a preference of purine-Gpurine sequences (purines are adenine and guanine) with bonding to the middle G. Cisplatin is a transition metal complex cis-diamine-dichloro-platinum and clinically used as anticancer drug. The effect of the drug is due to the ability to platinate the N-7 of guanine on the major groove site of DNA double helix. This chemical modification of platinum atom cross-links two adjacent guanines on the same DNA strand interfering with the mobility of DNA polymerases. Forces involved in DNA-drug recognition: Understanding the forces involved in the binding of proteins or small molecules to DNA is of prime importance due to two major reasons. Firstly, the design of sequence specific drugs having requisite affinity for DNA requires a knowledge how the structure of the drug is related to the specificity/affinity of binding and what structural modifications could result in a drug with desired qualities. Secondly, identifying the forces/energetics involved in such processes is fundamental to unraveling the mystery of molecular recognition in general and DNA binding in particular. Some of the forces that are known to contribute to biomolecular recognition and also to DNA-drug binding are direct electrostatic interactions, direct van der Waals/packing interactions, complex hydration/dehydration contributions composed of hydrophobic component, solvation electrostatics, solvation van der Waals, ion effects and entropy terms.
DNA-drug binding may be described in the following manner:
Consider DNA-drug binding in an aqueous environment. DNA is polyanionic in nature and the drug molecule is also often charged. The associated counterions lie near the charged groups and are also partially solvated. When binding occurs, it results in a displacement of solvent from the binding site on both the DNA and drug. Also, since there would be partial compensation of charges as the DNA and drug are oppositely charged, some counterions would be released into the bulk solvent and are solvated fully. Also, the binding process would be associated with some structural deformation/adaptation of the DNA as well as the drug molecule in order to accommodate each other. All these events are associated with some energetic gains/losses, the comprehensive estimation of which is a major challenge. We are attempting to understand the energetics of DNA-drug interaction by theoretically estimating the above contributions employing classical and statistical mechanical methods. Developing a theoretical protocol for detailed quantitative analysis of DNA-ligand binding in solution is a daunting task due to some major challenges. Simulations of DNA with solvent and the attendant counterion atmosphere require careful consideration to ensure system stability. Also, evolving a computationally efficient technique using statistical mechanical principles for quantitative estimates of binding free energies in large biomolecular systems is an equally challenging task. Our study is aimed at providing such a theoretical protocol for complementing experimental techniques and facilitating a minute study of the structure-energy relationships in DNA-drug complexes. Structural and conformational changes in the DNA and drug on binding in solution are associated with enthalpic and entropic contributions to the binding free energy, which can be theoretically estimated from ensembles of structures generated via simulations. The only drawback of this approach is the long time taken for the simulations. The other terms, namely, electrostatics, van der Waals, hydrophobic component, rotational and translational entropy can be estimated from single structures. The web tool, PreDDICTA, estimates the components of DNA-drug binding free energy which can be calculated from a single structure, and correlates it with experimental binding free energy and Tm, thus providing a swift method for evaluation of potential lead candidates for researchers pursuing structure based drug design for DNA. Chapter-21 Technique to study protein Introduction: Proteins are fundamental components of all living cells. They exhibit an enormous amount of chemical and structural diversity, enabling them to carry out an extraordinarily diverse range of biological functions. Scientists know that the critical feature of a protein is its ability to adopt the right shape for carrying out a particular function. But sometimes a protein twists into the wrong shape or has a missing part, preventing it from doing its job. Many diseases, such as Alzheimer's and "mad cow", are now known to result from proteins that have adopted an incorrect structure. Identifying a protein's shape, or structure, is a key to understanding its
biological function and its role in health and disease. Illuminating a protein's structure also paves the way for the development of new agents and devices to treat a disease. Yet solving the structure of a protein is no easy feat. It often takes scientists working in the laboratory months, sometimes years, to experimentally determine a single structure. Therefore, scientists have begun to turn toward computers to help predict the structure of a protein based on its sequence. The challenge lies in developing methods for accurately and reliably understanding this intricate relationship.
Determination of Protein Structure Traditionally, a protein's structure was determined using one of two techniques: 1.X-ray Crystallography. 2.Nuclear Magnetic Resonance (NMR) Spectroscopy. X-ray Crystallography . The basic building block of a crystal is called a unit cell. Each unit cell contains exactly one unique set of the crystal's components, the smallest possible set that is fully representative of the crystal. Crystals of a complex molecule, like a protein, produce a complex pattern of X-ray diffraction, or scattering of X-rays. When the crystal is placed in an X-ray beam, all of the unit cells present the same face to the beam; therefore, many molecules are in the same orientation with respect to the incoming X-rays. The X-ray beam enters the crystal and a number of smaller beams emerge: each one in a different direction, each one with a different intensity. If an X-ray detector, such as a piece of film, is placed on the opposite side of the crystal from the X-ray source, each diffracted ray, called a reflection, will produce a spot on the film. However, because only a few reflections can be detected with any one orientation of the crystal, an important component of any X-ray diffraction instrument is a device for accurately setting and changing the orientation of the crystal. The set of diffracted, emerging beams contains information about the underlying crystal structure. Nuclear Magnetic Resonance(NMR)Spectroscopy The basic phenomenon of NMR spectroscopy was discovered in 1945. In this technique, a sample is immersed in a magnetic field and bombarded with radio waves. These radio waves encourage the nuclei of the molecule to resonate, or spin. As the positively charged nucleus spins, the moving charge creates what is called a magnetic moment. The thermal motion of the moleculethe movement of the molecule associated with the temperature of the materialfurther creates a torque, or twisting force, that makes the magnetic moment "wobble" like a child's top. When the radio waves hit the spinning nuclei, they tilt even more, sometimes flipping over. These resonating nuclei emit a unique signal that is then picked up on a special radio receiver and translated using a decoder. This decoder is called the Fourier Transform algorithm, a complex equation that translates the language of the nuclei into something a scientist can understand. By measuring the frequencies at which different nuclei flip, scientists can determine molecular structure, as well as many other interesting properties of the molecule. In the past 10 years, NMR has proven to be a powerful alternative to X-ray crystallography for the determination of molecular structure. NMR has the advantage over crystallographic techniques in that experiments are performed in solution as opposed to a crystal lattice. However, the principles that make NMR possible tend to make this technique very time consuming and limit the application to small- and medium-sized molecules. Strategy behind NMR
Nuclear magnet resonance obtains the same high resolution using a very different strategy. NMR measures the distances between atomic nuclei, rather than the electron density in a molecule. With NMR, a strong, high frequency magnetic field stimulates atomic nuclei of the isotopes H-1, D-2, C-13, or N-15 (they have a magnetic spin) and measures the frequency of the magnetic field of the atomic nuclei during its oscillation period back to the initial state. The important step is to determine which resonance comes from which spin. The distance and type of neighboring nuclei determines the resonance frequency of the stimulated atomic nuclei. This dependence on next neighbors known as chemical shift (or spin-spin coupling constant) and reflects the local electronic environment and the information contained in 1-D NMR spectra. For proteins, NMR usually measures the spin of protons. The following reasons make the H-1 NMR spectroscopy the method of choice for biological macromolecules: - H is present at many sites in proteins, nucleic acids, and polysaccharides - H has a high abundance for each site - H nuclei is the most sensitive to detect
1-D spectra contain the information about all the chemical shifts of all the H in the protein. The frequency resolution is often not enough to distinguish individual chemical shifts. 2-D NMR solves these problems by containing information about the relative position of H in molecular structures. 2-D NMR spectra contain information about interaction between H that is covalently linked through one or two other atoms (COSY or correlation spectroscopy). Alternatively, pairs of H that can be close in space, even if they are from residues that are not close in sequence (NOE spectra, or Nuclear Overhauser Effect). A complete structure can thus be calculated by sequentially assigning cross peak correlations in 2-D spectra. Currently, the size limit for proteins amenable to NMR solution structure analysis is about 200 amino acids. An important feature of the identification of cross peaks is that regular patterns can be recognized that stem from secondary structure elements such as alpha helices and parallel or antiparallel beta sheets because they contain typical hydrogen bonding networks. NMR also requires the knowledge of the amino acid sequence, but the protein does not have to be in an ordered crystal, yet high concentrations of solubilized protein must be available (NMR structures are therefore also called solution structures). In biopolymers, the primary structure (sequence) logically breaks up the molecule into groups of coupled spins normally one or two groups per residue. This is true not only for proteins, but also for nucleic acids and polysaccharides. Fig. Observed NOEs in antiparallel and parallel sheets
Determination of membrane protein structure by rotational resonance NMR: bacteriorhodopsin. Rotationally resonant magnetization exchange, a new nuclear magnetic resonance (NMR) technique for measuring internuclear distances between like spins in solids, was used to determine the distance between the C-8 and C-18 carbons of retinal in two model compounds and in the membrane protein bacteriorhodopsin. Magnetization transfer between inequivalent spins with an isotropic shift separation, delta, is driven by magic angle spinning at a speed omega r that matches the rotational resonance condition delta = n omega r, where n is a small integer. The distances measured in this way for both the 6-s-cis- and 6-s-trans-retinoic acid model compounds agreed well with crystallographically known distances. In
bacteriorhodopsin the exchange trajectory between C-8 and C-18 was in good agreement with the internuclear distance for a 6-s-trans configuration [4.2 angstroms (A)] and inconsistent with that for a 6-s-cis configuration (3.1 A). The results illustrate that rotational resonance can be used for structural studies in membrane proteins and in other situations where diffraction and solution NMR techniques yield limited information. Structure of bacteriorhodopsin:
General: Current strategies for determining the structures of membrane proteins in lipid environments by NMR spectroscopy rely on the anisotropy of nuclear spin interactions, which are experimentally accessible through experiments performed on weakly and completely aligned samples. Importantly, the anisotropy of nuclear spin interactions results in a mapping of structure to the resonance frequencies and splittings observed in NMR spectra. Distinctive wheel-like patterns are observed in two-dimensional 1H-15N heteronuclear dipolar/15N chemical shift PISEMA (polarization inversion spin-exchange at the magic angle) spectra of helical membrane proteins in highly aligned lipid bilayer samples. One-dimensional dipolar waves are an extension of two-dimensional PISA (polarity index slant angle) wheels that map protein structures in NMR spectra of both weakly and completely aligned samples. Dipolar waves describe the periodic wave-like variations of the magnitudes of the heteronuclear dipolar couplings as a function of residue number in the absence of chemical shift effects. Since weakly aligned samples of proteins display these same effects, primarily as residual dipolar couplings, in solution NMR spectra, this represents a convergence of solid-state and solution NMR approaches to structure determination
X-ray crystallography and NMR are complementary techniques. NMR spectroscopy X-ray crystallography short time scale, protein folding long time scale, static structure solution, purity single crystal, purity < 20kD, domain any size, domain, complex functional active site active or inactive domains domains atomic nuclei, chemical bonds electron density resolution limit 2-3.5 resolution limit 2-3.5 primary structure must be known primary structure must be know (except if resolution is 2 or better for every single residue)
References: 1. Science 15 February 1991: Vol. 251. no. 4995, pp. 783 - 786 DOI: 10.1126/science.1990439 ARTICLE Determination of membrane protein structure by rotational resonance NMR: bacteriorhodopsin
F Creuzet, A McDermott, R Gebhard, K van der Hoef, MB Spijker-Assink, J Herzfeld, J Lugtenburg, MH Levitt, and RG Griffin. 2. Wikipedia.
3. Biochemistry by Lehninger, Chapter no.4: The three dimensional structure of Proteins, page no.116.
Chapter-22 Enzymology of DNA folding and unfolding WHAT IS DNA???
Deoxyribonucleic acid (DNA) is a nucleic acid that contains the genetic instructions used in the development and functioning of all known living organisms. The main role of DNA molecules is the long-term storage of information and DNA is often compared to a set of blueprints, since it contains the instructions needed to construct other components of cells, such as proteins and RNA molecules. The DNA segments that carry this genetic information are called genes, but other DNA sequences have structural purposes, or are involved in regulating the use of this genetic information. Chemically, DNA is a long polymer of simple units called nucleotides, with a backbone made of sugars and phosphate groups joined by ester bonds. Attached to each sugar is one of four types of molecules called bases. It is the sequence of these four bases along the backbone that encodes information. This information is read using the genetic code, which specifies the sequence of the amino acids within proteins. The code is read by copying stretches of DNA into the related nucleic acid RNA, in a process called transcription. Most of these RNA molecules are used to synthesize proteins, but others are used directly in structures such as ribosomes and spliceosomes. Within cells, DNA is organized into structures called chromosomes and the set of chromosomes within a cell make up a genome. These chromosomes are duplicated before cells divide, in a process called DNA replication. Eukaryotic organisms such as animals, plants, and fungi store their DNA inside the cell nucleus, while in prokaryotes such as bacteria it is found in the cell's cytoplasm. Within the chromosomes, chromatin proteins such as histones compact and organize DNA, which helps control its interactions with other proteins and thereby control which genes are transcribed.
DNA STRUCTURE
DNA is a long polymer made from repeating units called nucleotides.[1][2] The DNA chain is 22 to 26 ngstrms wide (2.2 to 2.6 nanometres), and one nucleotide unit is 3.3 ngstroms (0.33 nanometres) long.[3] Although each individual repeating
unit is very small, DNA polymers can be enormous molecules containing millions of nucleotides. For instance, the largest human chromosome, chromosome number 1, is 220 million base pairs long.[4] In living organisms, DNA does not usually exist as a single molecule, but instead as a tightly-associated pair of molecules.[5][6] These two long strands entwine like vines, in the shape of a double helix. The nucleotide repeats contain both the segment of the backbone of the molecule, which holds the chain together, and a base, which interacts with the other DNA strand in the helix. In general, a base linked to a sugar is called a nucleoside and a base linked to a sugar and one or more phosphate groups is called a nucleotide. If multiple nucleotides are linked together, as in DNA, this polymer is referred to as a polynucleotide.[7] The backbone of the DNA strand is made from alternating phosphate and sugar residues.[8] The sugar in DNA is 2-deoxyribose, which is a pentose (five carbon) sugar. The sugars are joined together by phosphate groups that form phosphodiester bonds between the third and fifth carbon atoms of adjacent sugar rings. These asymmetric bonds mean a strand of DNA has a direction. In a double helix the direction of the nucleotides in one strand is opposite to their direction in the other strand. This arrangement of DNA strands is called antiparallel. The asymmetric ends of DNA strands are referred to as the 5 (five prime) and 3 (three prime) ends. One of the major differences between DNA and RNA is the sugar, with 2-deoxyribose being replaced by the alternative pentose sugar ribose in RNA.[6] The DNA double helix is stabilized by hydrogen bonds between the bases attached to the two strands. The four bases found in DNA are adenine (abbreviated A), cytosine (C), guanine (G) and thymine (T). These four bases are shown below and are attached to the sugar/phosphate to form the complete nucleotide, as shown for adenosine monophosphate. These bases are classified into two types; adenine and guanine are fused five- and six-membered heterocyclic compounds called purines, while cytosine and thymine are six-membered rings called pyrimidines.[6] A fifth pyrimidine base, called uracil (U), usually takes the place of thymine in RNA and differs from thymine by lacking a methyl group on its ring. Uracil is not usually found in DNA, occurring only as a breakdown product of cytosine, but a very rare exception to this rule is a bacterial virus called PBS1 that contains uracil in its DNA.[9] In contrast, following synthesis of certain RNA molecules, a significant number of the uracils are converted to thymines by the enzymatic addition of the missing methyl group. This occurs mostly on structural and enzymatic RNAs like transfer RNAs and ribosomal RNA.[ 0] Major and minor grooves Animation of the structure of a section of DNA. The bases lie horizontally between the two spiraling strands. Large version[11] The double helix is a right-handed spiral. As the DNA strands wind around each other, they leave gaps between each set of phosphate backbones, revealing the sides of the bases inside (see animation). There are two of these grooves twisting around the surface of the double helix: one groove, the major groove, is 22 wide and the other, the minor groove, is 12 wide.[12] The narrowness of the minor groove means that the edges of the bases are more accessible in the major groove. As a result, proteins like transcription factors that can bind to specific sequences in double-stranded DNA usually make contacts to the sides of the bases
exposed in the major groove.[13]
Replication Cell division is essential for an organism to grow, but when a cell divides it must replicate the DNA in its genome so that the two daughter cells have the same genetic information as their parent. The double-stranded structure of DNA provides a simple mechanism for DNA replication. Here, the two strands are separated and then each strand's complementary DNA sequence is recreated by an enzyme called DNA polymerase. This enzyme makes the complementary strand by finding the correct base through complementary base pairing, and bonding it onto the original strand. As DNA polymerases can only extend a DNA strand in a 5 to 3 direction, different mechanisms are used to copy the antiparallel strands of the double helix.[71] In this way, the base on the old strand dictates which base appears on the new strand, and the cell ends up with a perfect copy of its DNA
MECHANISM OF DENATURATION AND RENATURATION OF DNA DNA DENATURATION : The two DNA strands, which form the double helix, are in opposite orientations (one runs in 5'-3'direction, the complementary in 3'-5'direction) and are connected by hydrogen bonds (see DNA structure). The rupture of these hydrogen bonds causes the DNA to separate or to "denature". Inside the cell, a partial separation of the DNA strands is necessary for replication and transcription and is caused by several proteins that use ATP. But in vitro the rupture of the hydrogen bonds can be caused by heat, alcaline conditions or chemical compounds The denaturation by heat is complete at 90C. Concerning alcalic conditions, a pH above 11.3 is able to denature DNA. In both cases, the process of denaturation is reversible. In the moment that the denaturating agent disappears, the DNA renaturalizes ( reassociates, reanneals) and it gets back to its native structures of a double helix. The temperature at which half of the molecules are denatured is called Tm (melting temperature). The Tm varies among organisms depending on their porcentage of G+C. A=T rich zones melt at lower temperatures than G=C rich zones, whereas GC rich zones tends to have a higher Tm. The content of G+C can therefore be calculated from the Tm value of the DNA. Also in biological systems, replication and transcription begins in the A=T rich regions. Denaturation at an elavated pH > 11 is caused by a rupture of the hydrogen bonds between the complementary bases. The alcalic conditions change the charges of the many side groups that are involved in the non-covalent binding between the bases.
Distilled water can also cause some degree of denaturation. It inhibits the neutralisation of the phosphate groups by salts like magnesium or sodium. The negative charge of the phopsphate group causes a repulsion and therefore a physical separation of the DNA strands. Chemical compounds like urea or formamide denature the DNA by directly reacting with the bases, thereby preventing normal base-pairing. The Tm is reduced proportionally to the concentration of denaturating chemical agents. If we decrease the concentration of either urea or formamide, DNA will renature again. Urea is used in denaturing gels to sequence DNA. Formaldehyde causes an irreversible denaturation, because it builds covalent bonds with the NH2 groups of the bases, thus preventing base-pairing. It can react with the DNA at room temperatures, because even in native DNA the bases are in a dynamic equilibrium of pairing and unpairing in discrete regions, called the respiration or palpitation of the DNA. It is thought that this phenomenon facilitates the access of regulatory proteins to the inner part of the DNA molecule. If DNA is to be kept in a denatured state, it needs to be transferred immediately from 100C to ice for fast cooling. This will prevent it from forming again the hydrogen bonds in an ordered manner. The denaturation of the DNA can be observed by a change of light absorption at a wavelength of 260 nm. Single stranded DNA has an appr. 40% higher absorption than double stranded DNA, since the heterocyclic rings of the bases absorb more light if they are not piled up or connected by hydrogen bonds insight the double helix. This phenomenon is called "hipercromicity". DNA RENATURATION :
The process of renaturation, observable as a change in light absorption, is a very valuable mechanism to evaluate the genetic relation between organisms. It shows the degree of similarity, correspondance and divergance between DNAs of different origin. A DNA that contains a high quantity of repetitive sequences (satellite sequences) will renaturate much faster than DNA that is mainly single sequences. Mice DNA for example renatures faster that bacterial DNA, even so it is 30 time bigger, because it contains many repetitive sequences. Shorter DNA molecules reassociate faster than longer molecules. In long molecules, many small fragments appear as a result of shering forces and the possibility that the complementary segments "find each other" is lower. This fact is valuable for example in compairing phage- to bacterial DNA. The renaturation occurs is two steps: 1.Two complementary sequences collide by incidence and they combine forming a helix structure. 2. The Base-pairing is extended over the whole strand in form of a "zipper". The process of renaturation is also called hybridization, and it can happen with its original complementary strand, but also with another sequence that has high homology but not complete. Hybridization is the basis of many molecular techniques studied in This course. What is Denaturation of DNA ?? As DNA is heated, it reaches a temperature where the strands seperate (DNA melts). The base pairs are separated as the H-bonds between them are broken and the strand unwind. This results in single stranded DNA.
It is possible to follow this process in a spectrophotometer, by observing the change in absorbance at 260nm. Unstacked bases (random orientation) absorb more light than neatly stacked (oriented) base-pairs. This results in a melting curve. The temperature at which DNA is half unfolded is called the melting temperature. Tm is a measure of the stability of DS-DNA under a given set of conditions. Stability, and therefore Tm, is affected by.... Base Composition - higher the GC content, the higher the Tm. Ionic Strength - as the ionic strength increases, so does Tm. Double helical DNA is stabilised by cations. Divalent cations (eg Mg2+) are more effective than monovalent cations (<NA+ or K+). Organic Solvents - formamide for instance lowers the Tm by weakening the hydrophobic interactions.
What is Reannealing of DNA ?? If melted DNA is cooled slowly, complementary strands will pair up again. This process is called "annealing". Only complementary strands can anneal. This is an important feature of DNA which is utilised in the laboratory when carrying out DNA hybridisation. Perfect annealing requires the perfect matching of base pairs, although mismatching occurs quite often. This gives less stable DNA, and can be monitored by watching the Tm. If the Tm lowers by 1 o, then 1% mismatch has occurred. The conditions of annealing, salt concentration and temperature, can determine the amount of mismatch that occurs. But it must be said that not all mismatching is bad. If, for instance, a gene was found in a rat, and researchers needed to know if a similiar gene was found in humans, then they would need to allow some mismatch, since the genetic sequences are unlikely to be exactly the same. "Stringency" is the term given to the conditions of annealing which control the extent of mismatching allowed. If the stringency of hybridisation is high, there is little or no mismatch with 95-100% of base-pairs matched correctly. At low stringency, only 40-50% of base-pairs are correctly repaired. Renaturation of DNA by a Saccharomyces cerevisiae Protein That Catalyzes Homologous Pairing and Strand Exchange* A protein from mitotic Saccharomyces cerevisiae cells that catalyzes homologous pairing and stranedx change was analyzed for the ability to catalyze other related reactions. The proteiwna s capable of renaturing complementary single-strandedD NA as evidenced by S1 nuclease assays and analysis of the reaction products by agarose gel electrophoresis and electron microscopy. Incubation of the yeast protewini th complementary single-strandedD NA resulted in the rapid formation ofl arge aggregates which did noetn ter agarose gels. These aggregates contained many branched structures consisting of both single-stranded and doublestranded DNA. These reactions required stoichiometric amounts of protein but shonwoe Ad TP requirement. The protein formed stable complexesw ith both single-stranded and double-stranded DNAs,h oeing a higher affinity for single-stranded DNA. The binding to single-stranded DNA resulted in the formation of
large protein:DNA aggregates. These aggregates were also formed in strand-exchange reactions and contained both substrate and product DNAs. Trheessuel ts demonstrate that the S. cerevisiae strand-exchange protein shares additional properties with the Escherichia coli recA protein which, by analogy, gives further indication that it might be implicated in homologous recombination
Mechanism of DNA denaturation in the presence of manganese ions
The unwinding of DNA strands in the presence of small concentrations of Mn2+ ions (2 10-4-4 10-4M) has been studied. The process of unwinding is nonequilibrium; the DNA strands are gradually unwound at a constant temperature corresponding to the beginning of the melting curve. There is no true renaturation in the partially melted DNA. It is shown in the paper that these effects are due to the aggregation of the unwound DNA regions. The Mn2+ ions are responsible for the binding of the unwound strands. The aggregation precludes renaturation, shifts the equilibrium towards the melted state, and causes slow unwinding at a constant temperature. The binding of denaturated regions seems to occur through the guanines
ADVANTAGES OF DENATURATION
1.PCR MELTING PROFILE(PCR MP)
The performance and convenience of a PCR melting profile (PCR MP) technique based on using low denaturation temperatures during ligation mediated PCR (LM PCR) of bacterial DNA is shown. We found that PCR MP technique is a rapid method that offers good discriminatory power, excellent reproducibility and may be applied for epidemiological studies. Results from strain genotyping illustrate that PCR MP is useful for the study of intraspecific genetic relatedness of strains and is as effective in discriminating closely related strains as the PFGE method, which is currently considered to be the gold standard for epidemiological studies. The usefulness of the PCR MP for molecular typing was shown for clinical strains of Escherichia coli, Enterococcus faecium VRE and Stapylococcus aureus. 2.HYBRIDIZATION OF DENATURED RNA AND SMALL DNA FRAGMENTS TRANSFERRED TO NITROCELLULOSE A simple and rapid method for transferring RNA from agarose gels to nitrocellulose paper for blot hybridization has been developed. Poly(A)+ and ribosomal RNAs transfer efficiently to nitrocellulose paper in high salt (3 M NaCl/0.3 M trisodium citrate) after denaturation with glyoxal and 50% (vol/vol) dimethyl sulfoxide. RNA also binds to nitrocellulose after treatment with methylmercuric hydroxide. The method is sensitive: about 50 pg of specific mRNA per band is readily detectable after hybridization with high specific activity probes (10(8) cpm/microgram). The RNA is stably bound to the nitrocellulose paper by this procedure, allowing removal of the hybridized probes and rehybridization of the RNA blots without loss of sensitivity. The use of nitrocellulose paper for the analysis of RNA by blot hybridization has several advantages over the use of activated paper (diazobenzyloxymethyl-paper). The method is simple, inexpensive, reproducible, and sensitive. In addition, denaturation of DNA with glyoxal and dimethyl sulfoxide promotes transfer and retention of small DNAs (100 nucleotides and larger) to nitrocellulose paper. A related method is also described for dotting RNA and DNA directly onto nitrocellulose paper treated with a high concentration of salt; under these conditions denatured DNA of less than 200 nucleotides is retained and hybridizes efficiently. 3.GENOMIC SEQUENCING Unique DNA sequences can be determined directly from mouse genomic DNA. A denaturing gel separates by size mixtures of unlabeled DNA fragments from complete restriction and partial chemical cleavages of the entire genome. These lanes of DNA are transferred and UV-crosslinked to nylon membranes. Hybridization with a short 32P-labeled single-stranded probe produces the image of a DNA sequence "ladder" extending from the 3' or 5' end of one restriction site in the genome. Numerous different sequences can be obtained from a single membrane by reprobing. Each band in these sequences represents 3 fg of DNA complementary to the probe. Sequence data from mouse immunoglobulin heavy chain genes from several cell types are presented. The genomic sequencing procedures are applicable to the analysis of genetic polymorphisms, DNA methylation at deoxycytidines, and nucleic acid-protein interactions at single nucleotide resolution
4.SUPERCOIL SEQUENCING:A FAST AND SIMPLE METHOD FOR SEQUENCING PLASMID DNA A method for obtaining sequence information directly from plasmid DNA is presented. The procedure involves the rapid preparation of clean supercoiled plasmid DNA from small bacterial cultures, its complete denaturation by alkali, and sequence determination using oligodeoxyribonucleotide-primed enzymatic DNA synthesis in the presence of dideoxynucleoside triphosphates. The advantages of the method include speed, simplicity, avoidance of additional cloning steps into single-stranded phage M13 vectors, and hence applicability to sequencing large numbers of samples. 5. HIGH EFFICIENCY TRANSFORMATION OF INTACT YEAST CELLS USING SINGLE STRANDED NUCLEIC ACIDS AS A CARRIER A method, using LiAc to yield competent cells, is described that increased the efficiency of genetic transformation of intact cells of Saccharomyces cerevisiae to more than 1 X 10(5) transformants per microgram of vector DNA and to 1.5% transformants per viable cell. The use of single stranded, or heat denaturated double stranded, nucleic acids as carrier resulted in about a 100 fold higher frequency of transformation with plasmids containing the 2 microns origin of replication. Single stranded DNA seems to be responsible for the effect since M13 single stranded DNA, as well as RNA, was effective. Boiled carrier DNA did not yield any increased transformation efficiency using spheroplast formation to induce DNA uptake, indicating a difference in the mechanism of transformation with the two methods. 6.DIDEOXY SEQUENCING METHOD USING DENATURED PLASMID TEMPLATES The dideoxy sequencing method in which denatured plasmid DNA is used as a template was improved. The method is simple and rapid: the recombinant plasmid DNA is extracted and purified by rapid alkaline lysis followed by ribonuclease treatment. The plasmid DNA is then immediately denatured with alkali and subjected to a sequencing reaction utilizing synthetic oligonucleotide primers. It takes only several hours from the start of the plasmid extraction to the end of the sequencing reaction. We examined each step of the procedure, and several points were found to be crucial for making the method reproducible and powerful: (i) the plasmid DNA should be free from RNA and open circular (or linear) DNA; (ii) a heptadecamer rather than a pentadecamer is recommended as a primer; and (iii) the sequencing reaction should be done at 37 degrees C or higher rather than at room temperature. The method enabled us to determine the sequence of more than a thousand nucleotides from a single template DNA. ADVANTAGES OF RENATURATION OF DNA 1.) Renaturation of DNA in the presence of ethidium bromide
The rate of renaturation of T2 DNA has been studied as a fuction of ethidium bound per nucleotide of denatured DNA. The Binding constants and number of binding sites for ethidium have been determined by spectral titration for denatured DNA at 55, 65, and 75C and for native DNA at 65C in 0.4M Na+. The rate of renaturation of T2 DNA was found to be independentof ethidium binding up to 0.03 moles per mole of nucleotide. Above 0.03 moles, the rate drops off precipitously approaching zero at 0.08 and 0.06 moles bound ethidium per nucleotide at 65C respectively. A study
was also made of the use of bound ethidium fluorescence as a probe for monitoring DNA renaturation reactions. 2) One hundred-fold acceleration of DNA renaturation rates in solution Solvents which accelerate DNA renaturation rates have been investigated. Addition of NaCl or LiCl to DNA in 2.4M Et4NCl initially increases renaturation rates at 45C and then leads to a loss of second-order behavior. The greatest accelerations are seen with LiCl and dilute DNA. Volume exclusion by dextran sulfate is the most effective method of accelerating DNA renaturation with concentrated DNA. Addition of dextran sulfate beyond 10-12% in 2.4M Et4NCl fails to increase the acceleration beyond approximately 10-fold. Accelerations of 100-fold may be achieved with 3540% dextran sulfate in 1M NaCl at 70C. No other mixed solvent system was found to be more effective, although acceleration may be achieved in solvents containing formamide or other denaturants. The acceleration in 2M NaCl occurs without loss of the normal concentration and temperature dependence of DNA renaturation and is also independent of dextran sulfate concentration if sufficient dextran sulfate is used. Dextran sulfate may be selectively precipitated by use of 1M CsCl. 3) Renaturation and Hybridization Studies of Mitochondrial DNA The products of the renaturation reaction of mitochondrial DNA from oocytes of Xenopus laevis have been studied by electron microscopy and CsCl equilibrium density gradient centrifugation. The reaction leads to the formation of intermediates containing single-stranded and double-stranded regions. Further reactions of these intermediates result in large complexes of interlinking doublestranded filaments. The formation of circular molecules of the same length as native circles of mitochondrial DNA was also observed. The formation of common high molecular weight complexes during joint reannealing of two DNA's with complementary sequences was used as a method to detect sequence homology in different DNA samples. Although this method does not produce quantitative data it offers several advantages in the present study. No homologies could be detected between the nuclear DNA and the mitochondrial DNA of X. laevis or of Rana pipiens. In interspecies comparisons homologies were found between the nuclear DNA's of X. laevis and the mouse and between the mitochondrial DNA's of X. laevis and the chick, but none between the mitochondrial DNA's of X. laevis and yeast. These results are interpreted as indicating the continuity of mitochondrial DNA during evolution.
Chapter-23
Safety and regulatory aspects of enzyme use
Introduction Only very few enzymes present hazards, because of their catalytic activity, to those handling them in normal circumstances but there are several areas of potential hazard arising from their chemical nature and source. These are allergenicity, activity-related toxicity, residual microbiological activity, and chemical toxicity. All enzymes, being proteins, are potential allergens and have especially potent effects if inhaled as a dust. Once an individual has developed an immune response as a result of inhalation or skin contact with the enzyme, re-exposure produces increasingly severe responses becoming dangerous or even fatal. Because of this, dry enzyme preparations have been replaced to a large extent by liquid preparations, sometimes deliberately made viscous to lower the likelihood of aerosol formation during handling. Where dry preparations must be used, as in the formulation of many enzyme detergents, allergenic responses by factory workers are a very significant problem particularly when fine-dusting powders are employed. Workers in such environments are usually screened for allergies and respiratory problems. The problem has been largely overcome by encapsulating and granulating dry enzyme preparations, a procedure that has been applied most successfully to the proteases and other enzymes used in detergents. Enzyme producers and users recognise that allergenicity will always be a potential problem and provide safety information concerning the handling of enzyme preparations. They stress that dust in the air should be avoided so weighing and manipulation of dry powders should be carried out in closed systems. Any spilt enzyme powder should be removed immediately, after first moistening it with water. Any waste enzyme powder should be dissolved in water before disposal into the sewage system. Enzyme on the skin or inhaled should be washed with plenty of water. Liquid preparations are inherently safer but it is important that any spilt enzyme is not allowed to dry as dust formation can then occur. The formation of aerosols (e.g. by poor operating procedures in centrifugation) must be avoided as these are at least as harmful as powders. Activity-related toxicity is much rarer but it must be remembered that proteases are potentially dangerous, particularly in concentrated forms and especially if inhaled. No enzyme has been found to be toxic, mutagenic or carcinogenic by itself as might be expected from its proteinaceous structure. However, enzyme preparations cannot be regarded as completely safe as such dangerous materials may be present as contaminants, derived from the enzyme source or produced during its processing or storage. The organisms used in the production of enzymes may themselves be sources of hazardous materials and have been the chief focus of attention by the regulatory authorities. In the USA, enzymes must be Generally Regarded As Safe (GRAS) by the FDA (Food and Drug Administration) in order to be used as a food ingredient. Such enzymes include -amylase,-amylase, bromelain, catalase, cellulase, ficin, galactosidase, glucoamylase, glucose isomerase, glucose oxidase, invertase, lactase, lipase, papain, pectinase, pepsin, rennet and trypsin. In the UK, the Food Additives and Contaminants Committee (FACC) of the Ministry of Agriculture, Fisheries and Food classified enzymes into five classes on the
basis of their safety for presence in the foods and use in their manufacture. Group A. Substances that the available evidence suggests are acceptable for use in food. Group B. Substances that on the available evidence may be regarded as provisionally acceptable for use in food but about which further information must be made available within a specified time for review. Group C. Substances for which the available evidence suggests toxicity and which ought not to be permitted for use in food until adequate evidence of their safety has been provided to establish their acceptability. Group D. Substances for which the available information indicates definite or probable toxicity and which ought not to be permitted for use in food. Group E. Substances for which inadequate or no toxicological data are available and for which it is not possible to express an opinion as to their acceptability for use in food. This classification takes into account the potential chemical toxicity from microbial secondary metabolites such as mycotoxins and aflotoxins. The growing body of knowledge on the long-term effects of exposure to these toxins is one of the major reasons for the tightening of legislative controls. The enzymes that fall into group A are exclusively plant and animal enzymes such as papain, catalase, lipase, rennet and various other proteases. Group B contains a very wide range of enzymes from microbial sources, many of which have been used in food or food processing for many hundreds of years. The Association of Microbial Food Enzyme Producers (AMFEP) has suggested subdivisions of the FACC's group B into: Class ain8 microorganisms that have traditionally been used in food or in food processing, including Bacillus subtilis, Aspergillus niger, Aspergillus oryzae, Rhizopus oryzae, Saccharomyces cerevisiae, Kluyveromyces fragilis, Kluyveromyces lactis and Mucor javanicus. Class bin8 microorganisms that are accepted as harmless contaminants present in food, including Bacillus stearothermophilus, Bacillus licheniformis, Bacillus coagulans, and Klebsiella in8aerogenes. Class cin8 microorganisms that are not included in Classes b and c, including Mucor miehei, Streptomyces albus, Trichoderma reesei, Actinoplanes missouriensis, and Penicillium emersonii. It was proposed that Class a should not be subjected to testing and that Classes b and c should be subjected to the following tests: acute oral toxicity in mice and rats, subacute oral toxicity for 4 weeks in rats, oral toxicity for 3 months in rats, and in vitro mutagenicity. In addition Class c should be tested for microorganism pathogenicity and, under exceptional circumstances, in vivo mutagenicity, teratogenicity, and carcinogenicity. The cost of the various tests needed to satisfy the legal requirements are very significant and must be considered during the determination of process costs. Plainly the introduction of an enzyme from a totally new source will be a very expensive matter. It may prove more satisfactory to clone such an enzyme into one of AMFEP's Class a organisms but this will first require new legislation to regulate the use of cloned microbes in foodstuffs. Some of the safety problems associated with the use of free enzymes may be overcome by using immobilised enzymes . This is an extremely safe technique, so long as the materials used are acceptable and neither they, nor the immobilised enzymes, leak into the product stream.
You might also like
- Notes Biomaterials IIDocument14 pagesNotes Biomaterials IIspartan100% (3)
- Radioactivity ProblemsDocument10 pagesRadioactivity Problemskarim adelNo ratings yet
- Bioanalytical Chemistry (CHEM311) - Lab ManualDocument29 pagesBioanalytical Chemistry (CHEM311) - Lab ManualSaravanan RajendrasozhanNo ratings yet
- Chapter Two: General Design ConsiderationsDocument27 pagesChapter Two: General Design ConsiderationsTeddy Ekubay GNo ratings yet
- Biochemistry - Final Exam Biology 020.305 December 15, 2011 Instructions For ExaminationDocument11 pagesBiochemistry - Final Exam Biology 020.305 December 15, 2011 Instructions For ExaminationJenna ReynoldsNo ratings yet
- Sample Exam 4 Fall 11Document14 pagesSample Exam 4 Fall 11janohxNo ratings yet
- AAC - Chapter 3 Centrifugation English - 20100325Document66 pagesAAC - Chapter 3 Centrifugation English - 20100325Sandeep Kumar100% (1)
- Protein FoldingDocument21 pagesProtein FoldingRONAK LASHKARINo ratings yet
- Separation of Lactose From Milk by UltrafiltrationDocument8 pagesSeparation of Lactose From Milk by UltrafiltrationLuisa FernandaNo ratings yet
- Analytical Chemistry EBT 253/3: Prepared By, Dr. Salmie Suhana Che AbdullahDocument12 pagesAnalytical Chemistry EBT 253/3: Prepared By, Dr. Salmie Suhana Che Abdullahmnfirdaus93100% (1)
- BIO307 Lecture 5 (Enzyme Kinetics I)Document11 pagesBIO307 Lecture 5 (Enzyme Kinetics I)Phenyo Mmereki100% (1)
- Ionic Liquids in Lipid Processing and Analysis: Opportunities and ChallengesFrom EverandIonic Liquids in Lipid Processing and Analysis: Opportunities and ChallengesXuebing XuNo ratings yet
- Chap9 Downstream ProcessingDocument25 pagesChap9 Downstream ProcessingsadatrafiaNo ratings yet
- Liquid Liquid ExtractionDocument20 pagesLiquid Liquid ExtractionGitanjali SahuNo ratings yet
- (Springer Laboratory) Prof. Sadao Mori, Dr. Howard G. Barth (Auth.) - Size Exclusion Chromatography-Springer-Verlag Berlin Heidelberg (1999)Document235 pages(Springer Laboratory) Prof. Sadao Mori, Dr. Howard G. Barth (Auth.) - Size Exclusion Chromatography-Springer-Verlag Berlin Heidelberg (1999)Luis Paulo BernardiNo ratings yet
- Gel Filteration ChromatographyDocument6 pagesGel Filteration ChromatographyAzka AsimNo ratings yet
- Lecture Notes-Bioreactor Design and Operation-1Document19 pagesLecture Notes-Bioreactor Design and Operation-1LCtey100% (1)
- Scale Up FermentersDocument11 pagesScale Up FermentersLouella100% (1)
- Lab Manual For DSPDocument44 pagesLab Manual For DSPaathiraNo ratings yet
- Lecture 1 CatalysisDocument28 pagesLecture 1 CatalysisMo MobarkNo ratings yet
- Biosepartaion Engineering: Ch.1: Bioseparation & Biological MaterialsDocument89 pagesBiosepartaion Engineering: Ch.1: Bioseparation & Biological MaterialsAlex MaximusNo ratings yet
- 14Document110 pages14Meyy SarrahNo ratings yet
- Class Notes Optical IsomerismDocument9 pagesClass Notes Optical IsomerismDeepanshu 1459No ratings yet
- In Silico Molecular Docking of Marine Drugs Against Cancer ProteinsDocument5 pagesIn Silico Molecular Docking of Marine Drugs Against Cancer ProteinsSEP-PublisherNo ratings yet
- Synthesis and Property Comparison of Silicone PolymersDocument3 pagesSynthesis and Property Comparison of Silicone PolymersDaniel RodmanNo ratings yet
- Biomaterials Exam 2 HW Answer Key PDFDocument24 pagesBiomaterials Exam 2 HW Answer Key PDFChelle VillasisNo ratings yet
- Department of Chemistry GC Women University, Sialkot: Name: Roll No.Document2 pagesDepartment of Chemistry GC Women University, Sialkot: Name: Roll No.Esha Rani Waheed100% (1)
- Stoichiometry of Microbial Growth and Product FormationDocument16 pagesStoichiometry of Microbial Growth and Product FormationAmiel DionisioNo ratings yet
- Chapter 2 Introduction To MaterialsDocument47 pagesChapter 2 Introduction To MaterialsDDVANNo ratings yet
- Intercalation Compounds of Graphite PDFDocument186 pagesIntercalation Compounds of Graphite PDFchanchan88vn100% (1)
- Protein Based NanostructuresDocument3 pagesProtein Based NanostructuresDannyMarlonJ100% (1)
- Materials Science & Engineering Introductory E-BookDocument13 pagesMaterials Science & Engineering Introductory E-BookSmitha KollerahithluNo ratings yet
- Lecture 01-03Document15 pagesLecture 01-03Fayaz AhmedNo ratings yet
- Bio Inorganic 1 PPT ChemistryDocument57 pagesBio Inorganic 1 PPT ChemistryShantanu MawaskarNo ratings yet
- Quiz On Enzyme KineticsDocument3 pagesQuiz On Enzyme KineticsHens Christian FuentesNo ratings yet
- Lipid and Amino Acid Metabolism: Chapter ObjectivesDocument11 pagesLipid and Amino Acid Metabolism: Chapter ObjectivesLarry411100% (1)
- Answer HPLCDocument3 pagesAnswer HPLCMuhammad Firdaus100% (1)
- Chapter 2 Water: Multiple Choice QuestionsDocument10 pagesChapter 2 Water: Multiple Choice QuestionsFrank WuNo ratings yet
- Centrifugation: NotesDocument13 pagesCentrifugation: Notesprism1702100% (1)
- C H O + A O + B NH C C H NO + D H O+eCO: InstructionsDocument4 pagesC H O + A O + B NH C C H NO + D H O+eCO: InstructionsJohn Paul Jandayan33% (3)
- BIOC 307 Old Exam 1Document17 pagesBIOC 307 Old Exam 1Katie RoseNo ratings yet
- Chapter 1 Introduction To Biochemistry and ThermodynamicsDocument4 pagesChapter 1 Introduction To Biochemistry and ThermodynamicsMamamiaNo ratings yet
- Lectura - EnzimasDocument13 pagesLectura - EnzimasMirella Bravo Benites0% (1)
- CHE 311 CHE Calculations IDocument4 pagesCHE 311 CHE Calculations IMikho SaligueNo ratings yet
- Crystallization and Its Application in Pharmaceutical Field PDFDocument10 pagesCrystallization and Its Application in Pharmaceutical Field PDFApollo RoabNo ratings yet
- CHE 46: Biochemical EngineeringDocument30 pagesCHE 46: Biochemical EngineeringJunn Edgar LibotNo ratings yet
- Rr212305-Chemical and BiothermodynamicsDocument1 pageRr212305-Chemical and BiothermodynamicssivabharathamurthyNo ratings yet
- Manual-4 6 7Document412 pagesManual-4 6 7Nina Brown100% (1)
- Nzymes: By: Mrs. Kalaivani Sathish. M. Pharm, Assistant Professor, Pims - PanipatDocument63 pagesNzymes: By: Mrs. Kalaivani Sathish. M. Pharm, Assistant Professor, Pims - Panipaturmila pandeyNo ratings yet
- Lecture Notes On Separation of Stable IsotopesDocument31 pagesLecture Notes On Separation of Stable IsotopesKaranam.Ramakumar100% (1)
- Transition Metal ComplexesDocument103 pagesTransition Metal Complexesidownloadbooksforstu100% (1)
- 05Document18 pages05gatototNo ratings yet
- CHAPTER 3 Phase Diagram TTT HT - 1stDocument25 pagesCHAPTER 3 Phase Diagram TTT HT - 1stAriff AziziNo ratings yet
- University Life Purpose: VisionDocument7 pagesUniversity Life Purpose: VisionMaria Cecille Sarmiento GarciaNo ratings yet
- BIO307 Lecture 3 (Introduction To Enzymes)Document13 pagesBIO307 Lecture 3 (Introduction To Enzymes)Phenyo MmerekiNo ratings yet
- Degradation of Methylene Blue Dye Using A Photochemical ReactorDocument5 pagesDegradation of Methylene Blue Dye Using A Photochemical ReactorIJSTENo ratings yet
- Oscillometry and Conductometry: International Series of Monographs on Analytical ChemistryFrom EverandOscillometry and Conductometry: International Series of Monographs on Analytical ChemistryNo ratings yet
- 1-2 Introduction BiotechDocument20 pages1-2 Introduction BiotechBT20CME033 Gautam TahilyaniNo ratings yet
- 32721Document33 pages32721prasadbheemNo ratings yet
- Bty364 - Fundamentals of Food MicrobiologyDocument7 pagesBty364 - Fundamentals of Food MicrobiologysmbhattNo ratings yet
- 4 26Document28 pages4 26smbhattNo ratings yet
- Class 'B' Posts. Application Form, Academic Performance Indicator (API) Based Performance Based AppraisalDocument20 pagesClass 'B' Posts. Application Form, Academic Performance Indicator (API) Based Performance Based AppraisalsmbhattNo ratings yet
- Aw Food SafetyDocument9 pagesAw Food SafetyBiodiesel Pala FredricksenNo ratings yet
- Animal Cell Culture S M BhattDocument1 pageAnimal Cell Culture S M BhattsmbhattNo ratings yet
- Allostery in HemoglobiinDocument4 pagesAllostery in HemoglobiinsmbhattNo ratings yet
- Light MicroscopeDocument1 pageLight MicroscopesmbhattNo ratings yet