You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Tamil Ilakkanam Books For TNPSCDocument113 pagesTamil Ilakkanam Books For TNPSCkk_kamalakkannan100% (1)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Sickle Cell AnemiaDocument13 pagesSickle Cell Anemiamayra100% (1)
- PsychFirstAidSchools PDFDocument186 pagesPsychFirstAidSchools PDFAna ChicasNo ratings yet
- Osce Pro III Trial AnswerDocument69 pagesOsce Pro III Trial AnswerDr Onn Azli Puade100% (3)
- Got GottmanDocument3 pagesGot GottmanaalcantaraNo ratings yet
- Republic Vs CA - Land Titles and Deeds - GR No. 127060Document3 pagesRepublic Vs CA - Land Titles and Deeds - GR No. 127060Maya Sin-ot ApaliasNo ratings yet
- Brain and LanguageDocument3 pagesBrain and LanguageJasper AngelesNo ratings yet
- MTAP Math ChallengeDocument5 pagesMTAP Math ChallengeHaron Abedin100% (1)
- Case Report Example - GynaecologyDocument14 pagesCase Report Example - GynaecologyDr Onn Azli PuadeNo ratings yet
- Viva Borderline Year 2012/2013 QuestionDocument1 pageViva Borderline Year 2012/2013 QuestionDr Onn Azli PuadeNo ratings yet
- Osce Pro III Trial QuestionDocument19 pagesOsce Pro III Trial QuestionDr Onn Azli PuadeNo ratings yet
- Practice QUestion MEQ 1Document4 pagesPractice QUestion MEQ 1Dr Onn Azli PuadeNo ratings yet
- Clinical Case For PRO 3 - 2011/2012 BatchDocument3 pagesClinical Case For PRO 3 - 2011/2012 BatchDr Onn Azli PuadeNo ratings yet
- Family Medicine LogbookDocument2 pagesFamily Medicine LogbookDr Onn Azli PuadeNo ratings yet
- Answer Trial RadioDocument47 pagesAnswer Trial RadioDr Onn Azli Puade100% (1)
- Musculoskeletal BlockDocument3 pagesMusculoskeletal BlockDr Onn Azli PuadeNo ratings yet
- Nervous System BlockDocument2 pagesNervous System BlockDr Onn Azli PuadeNo ratings yet
- Respiratory BlockhantarDocument3 pagesRespiratory BlockhantarDr Onn Azli PuadeNo ratings yet
- Genitourinary BlockDocument2 pagesGenitourinary BlockDr Onn Azli PuadeNo ratings yet
- Haematological BlockDocument2 pagesHaematological BlockDr Onn Azli PuadeNo ratings yet
- Cardiovascular BlockhantarDocument4 pagesCardiovascular BlockhantarDr Onn Azli PuadeNo ratings yet
- Assessment 1Document1 pageAssessment 1Dr Onn Azli PuadeNo ratings yet
- Endocrine BlockDocument2 pagesEndocrine BlockDr Onn Azli PuadeNo ratings yet
- Genitourinary BlockDocument3 pagesGenitourinary BlockDr Onn Azli PuadeNo ratings yet
- Gastrointestinal BlockhantarDocument3 pagesGastrointestinal BlockhantarDr Onn Azli PuadeNo ratings yet
- Case 1Document15 pagesCase 1Dr Onn Azli PuadeNo ratings yet
- Reproductive Block1Document2 pagesReproductive Block1Dr Onn Azli PuadeNo ratings yet
- Assessment 2Document14 pagesAssessment 2Dr Onn Azli PuadeNo ratings yet
- Assesment Selanjar 1Document10 pagesAssesment Selanjar 1Dr Onn Azli PuadeNo ratings yet
- Nervous System BlockDocument2 pagesNervous System BlockDr Onn Azli PuadeNo ratings yet
- Genitourinary BlockDocument2 pagesGenitourinary BlockDr Onn Azli PuadeNo ratings yet
- Respiratory BlockhantarDocument3 pagesRespiratory BlockhantarDr Onn Azli PuadeNo ratings yet
- Haematological BlockDocument2 pagesHaematological BlockDr Onn Azli PuadeNo ratings yet
- Musculoskeletal BlockDocument3 pagesMusculoskeletal BlockDr Onn Azli PuadeNo ratings yet
- Genitourinary BlockDocument3 pagesGenitourinary BlockDr Onn Azli PuadeNo ratings yet
- Gastrointestinal BlockhantarDocument3 pagesGastrointestinal BlockhantarDr Onn Azli PuadeNo ratings yet
- Chapter Three: Research MethodologyDocument3 pagesChapter Three: Research MethodologyEng Abdulkadir MahamedNo ratings yet
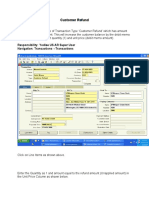
- Customer Refund: Responsibility: Yodlee US AR Super User Navigation: Transactions TransactionsDocument12 pagesCustomer Refund: Responsibility: Yodlee US AR Super User Navigation: Transactions TransactionsAziz KhanNo ratings yet
- Biblehub Com Commentaries Matthew 3 17 HTMDocument21 pagesBiblehub Com Commentaries Matthew 3 17 HTMSorin TrimbitasNo ratings yet
- Mathematics in The Primary Curriculum: Uncorrected Proof - For Lecturer Review OnlyDocument12 pagesMathematics in The Primary Curriculum: Uncorrected Proof - For Lecturer Review OnlyYekeen Luqman LanreNo ratings yet
- Oleracea Contain 13.2% Dry Matter, 15.7% Crude Protein, 5.4% Ether ExtractionDocument47 pagesOleracea Contain 13.2% Dry Matter, 15.7% Crude Protein, 5.4% Ether ExtractionJakin Aia TapanganNo ratings yet
- Sri Guru Parampara Stotram CompressDocument14 pagesSri Guru Parampara Stotram CompressSatishPavurayalaNo ratings yet
- Lesson I. Background InformationDocument21 pagesLesson I. Background InformationsuidivoNo ratings yet
- Man Is Made by His BeliefDocument2 pagesMan Is Made by His BeliefLisa KireechevaNo ratings yet
- Impact of Micro FinanceDocument61 pagesImpact of Micro FinancePerry Arcilla SerapioNo ratings yet
- Software Project Management PDFDocument125 pagesSoftware Project Management PDFUmirNo ratings yet
- VestibuleDocument17 pagesVestibuleDeepti MangalNo ratings yet
- IUGRDocument4 pagesIUGRMichael Spica RampangileiNo ratings yet
- Confidential Recommendation Letter SampleDocument1 pageConfidential Recommendation Letter SamplearcanerkNo ratings yet
- Asian Journal of ForestryDocument3 pagesAsian Journal of ForestryTeguh MuslimNo ratings yet
- Openfire XXMPP Server On Windows Server 2012 R2Document9 pagesOpenfire XXMPP Server On Windows Server 2012 R2crobertoNo ratings yet
- Villegas vs. Subido - Case DigestDocument5 pagesVillegas vs. Subido - Case DigestLouvanne Jessa Orzales BesingaNo ratings yet
- International Human Rights LawDocument21 pagesInternational Human Rights LawRea Nica GeronaNo ratings yet
- Angel Number 1208 Meaning Increased FaithDocument1 pageAngel Number 1208 Meaning Increased FaithKhally KatieNo ratings yet
- 1sebastian Vs CalisDocument6 pages1sebastian Vs CalisRai-chan Junior ÜNo ratings yet
- Soal Midtest + Kunci JawabanDocument28 pagesSoal Midtest + Kunci JawabanYuyun RasulongNo ratings yet
- SAP CRM Tax ConfigurationDocument18 pagesSAP CRM Tax Configurationtushar_kansaraNo ratings yet
- Pakistan List of Approved Panel PhysicianssDocument5 pagesPakistan List of Approved Panel PhysicianssGulzar Ahmad RawnNo ratings yet
- Measure For Measure AngeloDocument1 pageMeasure For Measure AngeloRoger Knight100% (1)