You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Unity Game Development ScriptingDocument27 pagesUnity Game Development ScriptingChang Jae Lee0% (1)
- Electron Delocalization and Molecular StructureDocument3 pagesElectron Delocalization and Molecular Structurehussai7No ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Group 7 The Halogens KLASSDocument12 pagesGroup 7 The Halogens KLASSKimberly LinderholmNo ratings yet
- Chemistry HydrocarbonsDocument144 pagesChemistry HydrocarbonsRudensNo ratings yet
- Bloch's Theorem and Krönig-Penney ModelDocument30 pagesBloch's Theorem and Krönig-Penney ModelChang Jae Lee50% (2)
- Ulangkaji Akhir Menjelang SPM AnswerDocument36 pagesUlangkaji Akhir Menjelang SPM AnswerHee Ting Wong100% (1)
- CHE 025 1st Periodic Exam ReviewDocument4 pagesCHE 025 1st Periodic Exam ReviewCelina PilloraNo ratings yet
- Edited-PhysicalSci12 Q1 Mod1 Week1 Formation of Elements v3Document21 pagesEdited-PhysicalSci12 Q1 Mod1 Week1 Formation of Elements v3Rogielyn P. Capin64% (11)
- Optical PropertiesDocument115 pagesOptical PropertiesRoss JonesNo ratings yet
- Stem Escape Room: Leading Up To The EventDocument18 pagesStem Escape Room: Leading Up To The EventChang Jae LeeNo ratings yet
- Heliyon: Jesús S Anchez-Martín, Mario Corrales-Serrano, Amalia Luque-Sendra, Francisco Zamora-PoloDocument11 pagesHeliyon: Jesús S Anchez-Martín, Mario Corrales-Serrano, Amalia Luque-Sendra, Francisco Zamora-PoloChang Jae LeeNo ratings yet
- Two Dimensional Electron Gas, Quantum Wells & Semiconductor SuperlatticesDocument41 pagesTwo Dimensional Electron Gas, Quantum Wells & Semiconductor SuperlatticesChang Jae LeeNo ratings yet
- Unity Cutscene TutDocument10 pagesUnity Cutscene TutChang Jae LeeNo ratings yet
- Chemical PseudopottentialDocument24 pagesChemical PseudopottentialChang Jae LeeNo ratings yet
- Update On Type II Diabetes in America - The Reasoned SocietyDocument7 pagesUpdate On Type II Diabetes in America - The Reasoned SocietyChang Jae LeeNo ratings yet
- Escape the Lab - Solve the Mystery of the Periodic TableDocument31 pagesEscape the Lab - Solve the Mystery of the Periodic TableChang Jae LeeNo ratings yet
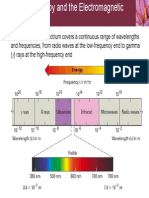
- 10.5 Spectroscopy and The Electromagnetic SpectrumDocument22 pages10.5 Spectroscopy and The Electromagnetic SpectrumChang Jae LeeNo ratings yet
- Local Manipulation of Nuclear Spin in A Semiconductor Quantum WellDocument18 pagesLocal Manipulation of Nuclear Spin in A Semiconductor Quantum WellChang Jae LeeNo ratings yet
- Symbolic Methods and Software Overview For Lie Symmetry AnalysisDocument60 pagesSymbolic Methods and Software Overview For Lie Symmetry AnalysisChang Jae LeeNo ratings yet
- Kronig Penney ModelDocument10 pagesKronig Penney ModelPhạm Hầu Thanh ViệtNo ratings yet
- Van Hove SingularityDocument3 pagesVan Hove SingularityChang Jae LeeNo ratings yet
- Numerical DifferentiationDocument2 pagesNumerical DifferentiationChang Jae LeeNo ratings yet
- PVT Relationships Real Gases Computational LabDocument6 pagesPVT Relationships Real Gases Computational LabChang Jae LeeNo ratings yet
- Many-Body Fermion Density MatricesDocument1,278 pagesMany-Body Fermion Density MatricesChang Jae LeeNo ratings yet
- Integrate The Feynman WayDocument6 pagesIntegrate The Feynman WayEl RuloNo ratings yet
- Quantum Jump Statatistics in Cavity JKPSDocument16 pagesQuantum Jump Statatistics in Cavity JKPSChang Jae LeeNo ratings yet
- DFT Solid State IntroductionDocument34 pagesDFT Solid State IntroductionChang Jae LeeNo ratings yet
- Lecture Note On Solid State PhysicsDocument38 pagesLecture Note On Solid State PhysicsChang Jae LeeNo ratings yet
- Reciprocal Space and Brillouin ZonesDocument10 pagesReciprocal Space and Brillouin ZonesChang Jae LeeNo ratings yet
- Shockwace Application To Scientific TeachingDocument9 pagesShockwace Application To Scientific TeachingChang Jae LeeNo ratings yet
- Plane Waves and KDocument21 pagesPlane Waves and KChang Jae LeeNo ratings yet
- DFT in PracticeDocument33 pagesDFT in PracticeChang Jae LeeNo ratings yet
- Semiconductor Defect ClassificationDocument6 pagesSemiconductor Defect ClassificationChang Jae LeeNo ratings yet
- Solid State PhysicsDocument281 pagesSolid State PhysicsChang Jae LeeNo ratings yet
- Basics of DFT Applications To Solids and SurfacesDocument17 pagesBasics of DFT Applications To Solids and SurfacesChang Jae LeeNo ratings yet
- Semester V Paper Code ETME 305 L4 T0 C4Document26 pagesSemester V Paper Code ETME 305 L4 T0 C4Amit JangraNo ratings yet
- MIT 5.112 Fall 2011 SyllabusDocument2 pagesMIT 5.112 Fall 2011 SyllabusbitternessinmymouthNo ratings yet
- Introduction To Molecular Orbital TheoryDocument17 pagesIntroduction To Molecular Orbital TheoryGeoorge VouyiouklakisNo ratings yet
- Gemma C. Solomon Et Al - Exploring Local Currents in Molecular JunctionsDocument6 pagesGemma C. Solomon Et Al - Exploring Local Currents in Molecular JunctionsGomsajNo ratings yet
- General Chemistry CHEM 1012 Chapter 1 An PDFDocument113 pagesGeneral Chemistry CHEM 1012 Chapter 1 An PDFmezgebu biresawNo ratings yet
- Chemistry-Part Test-2 XiiiDocument7 pagesChemistry-Part Test-2 XiiiRaju SinghNo ratings yet
- Chemistry 1311 Problem Set 1Document5 pagesChemistry 1311 Problem Set 1qabusalemNo ratings yet
- OxfordAQA 9620 CH02 WRE Jun22 v1.0Document5 pagesOxfordAQA 9620 CH02 WRE Jun22 v1.0MirandaxxNo ratings yet
- Common Organic Compounds: Properties and Uses: Redeveloped Division Initiated Self-Learning ModuleDocument21 pagesCommon Organic Compounds: Properties and Uses: Redeveloped Division Initiated Self-Learning ModuleMaribeth Q. AdierNo ratings yet
- Corma 1985Document124 pagesCorma 1985Leonan Dos Santos RodriguesNo ratings yet
- Module 3 Q2 Gen Chem IIDocument10 pagesModule 3 Q2 Gen Chem IIMengieNo ratings yet
- Mark Scheme Periodic Table Elements and Physical ChemistryDocument25 pagesMark Scheme Periodic Table Elements and Physical ChemistryEsam ELNOAMANYNo ratings yet
- SCI02-Prelims The Big Bang Theory & NucleosynthesisDocument12 pagesSCI02-Prelims The Big Bang Theory & NucleosynthesisMORAN DianaNo ratings yet
- Chemistry Water NotesDocument6 pagesChemistry Water NotesAzaz AhmedNo ratings yet
- A New Family of Anisotropic Spin Glasses-Ba1-xLa1+xMnO4+δ PDFDocument226 pagesA New Family of Anisotropic Spin Glasses-Ba1-xLa1+xMnO4+δ PDFjacoboNo ratings yet
- Alexander M. Spokoyny Et Al - Carborane-Based Pincers: Synthesis and Structure of SeBSe and SBS PD (II) ComplexesDocument2 pagesAlexander M. Spokoyny Et Al - Carborane-Based Pincers: Synthesis and Structure of SeBSe and SBS PD (II) ComplexesGomsajNo ratings yet
- Inorganica Chimica Acta: SciencedirectDocument7 pagesInorganica Chimica Acta: SciencedirectlianyNo ratings yet
- Infrared Spectra Alteration in Water Proximate ToDocument14 pagesInfrared Spectra Alteration in Water Proximate ToLarita NievasNo ratings yet
- Adobe Scan 06-Mar-2022 PDFDocument18 pagesAdobe Scan 06-Mar-2022 PDFCerena SinghNo ratings yet
- Chapter 1 Ncert SolutionsDocument22 pagesChapter 1 Ncert SolutionsHarsh MahalwarNo ratings yet
- Lesson 2 Organic Chemistry ConceptsDocument37 pagesLesson 2 Organic Chemistry Conceptsnorlene narita macedaNo ratings yet
- Sample Questions - Chapter 6Document4 pagesSample Questions - Chapter 6Rasel IslamNo ratings yet
- Introduction To Materials Tutorial 2Document11 pagesIntroduction To Materials Tutorial 2Azoz BubiNo ratings yet
- Chemistry: Presented By: Mrs. Marie Nella T. VictoriaDocument75 pagesChemistry: Presented By: Mrs. Marie Nella T. VictoriaJESPHER GARCIANo ratings yet