You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Pharmaco-Complexity: Anthony J. Hickey Hugh D.C. SmythDocument77 pagesPharmaco-Complexity: Anthony J. Hickey Hugh D.C. SmythAbdo EnnarhmouchyNo ratings yet
- MVRS Man en PDFDocument36 pagesMVRS Man en PDFelmer sanchez0% (1)
- FCC MANUAL 5-FCC Catalyst AnalysisDocument11 pagesFCC MANUAL 5-FCC Catalyst AnalysisshanpyanNo ratings yet
- 2005 Crude Oil Emulsions - A State-Of-The-Art ReviewDocument9 pages2005 Crude Oil Emulsions - A State-Of-The-Art ReviewOscar RoaNo ratings yet
- Acid Gas TreatingDocument28 pagesAcid Gas TreatingFaisal NadeemNo ratings yet
- Sorbent Cost and Performance in CO2 Capture SystemsDocument5 pagesSorbent Cost and Performance in CO2 Capture SystemsserchNo ratings yet
- Oil and Fat Assignment PDFDocument21 pagesOil and Fat Assignment PDFM Asrar SidonNo ratings yet
- Phenol Removal From Aqueous System by Jute StickDocument4 pagesPhenol Removal From Aqueous System by Jute StickAdnan Ahmed ChahalNo ratings yet
- Surface Chemistry of Solid Catalysts PDFDocument37 pagesSurface Chemistry of Solid Catalysts PDFCorby TranNo ratings yet
- Preparative Separation and Purification of Lycopene From Tomato Skins Extracts by Macroporous Adsorption ResinsDocument8 pagesPreparative Separation and Purification of Lycopene From Tomato Skins Extracts by Macroporous Adsorption ResinsТетяна КозицькаNo ratings yet
- Abdallah 2020Document10 pagesAbdallah 2020Piotr KucharskiNo ratings yet
- Definition of Mass TransferDocument6 pagesDefinition of Mass TransferBruno Lupi Barroso50% (2)
- Purification of Anthocyanins From Red Cabbage Using Semi Interpenetrating Network Hydrogel Beads in A Packed Bed ColumnDocument9 pagesPurification of Anthocyanins From Red Cabbage Using Semi Interpenetrating Network Hydrogel Beads in A Packed Bed ColumnShankara NarayananNo ratings yet
- Utility of Immobilized Recombinant Carbonic Anhydrase of Bacillus Halodurans TSLV1 On The Surface of Modified Iron Magnetic Nanoparticles in Carbon SequestrationDocument8 pagesUtility of Immobilized Recombinant Carbonic Anhydrase of Bacillus Halodurans TSLV1 On The Surface of Modified Iron Magnetic Nanoparticles in Carbon SequestrationHimadri BoseNo ratings yet
- Kinetika 3 CastellanDocument47 pagesKinetika 3 CastellanledikimetzeronaNo ratings yet
- Separation Note (CHE 461)Document32 pagesSeparation Note (CHE 461)JimNo ratings yet
- Sip Sample StudyDocument19 pagesSip Sample StudyGojo SatoruNo ratings yet
- Virus Capture and Destruction by LabelFree Graphene Oxide For Detection and Disinfection ApplicationsDocument6 pagesVirus Capture and Destruction by LabelFree Graphene Oxide For Detection and Disinfection ApplicationsFrancisco José Fontelles ObelenisNo ratings yet
- CHY1009 Chemistry and Environmental Studies - SyllabusDocument3 pagesCHY1009 Chemistry and Environmental Studies - SyllabusKrishna Kamal PeddiboinaNo ratings yet
- Use of Papaya Seeds As A Biosorbent of MDocument9 pagesUse of Papaya Seeds As A Biosorbent of MAbdulrahmanNo ratings yet
- Chemical Engineering ResourcesDocument227 pagesChemical Engineering Resourcesdvtherion100% (2)
- Methods of Ammonia Removal in Anaerobic Digestion: A Review: Niclas Krakat, Burak Demirel, Reshma Anjum and Donna DietzDocument14 pagesMethods of Ammonia Removal in Anaerobic Digestion: A Review: Niclas Krakat, Burak Demirel, Reshma Anjum and Donna Dietzengr_afsoomro3147No ratings yet
- Materials For Separation Membranes in HydrogenDocument16 pagesMaterials For Separation Membranes in HydrogenIbrahim Al-MutazNo ratings yet
- Orientation ReportDocument43 pagesOrientation ReportRyan eqbalNo ratings yet
- Rr410802 Chemical Reaction Engineering IIDocument8 pagesRr410802 Chemical Reaction Engineering IISrinivasa Rao G100% (3)
- PolymersDocument17 pagesPolymersGhirlie Eunice LopezNo ratings yet
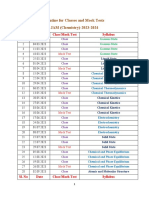
- Routine For JAM - 2023-2024Document2 pagesRoutine For JAM - 2023-2024Rahul NathNo ratings yet
- A Review of Chemicals To Produce Activated Carbon From Agricultural Waste BiomassDocument35 pagesA Review of Chemicals To Produce Activated Carbon From Agricultural Waste BiomassKeralem MuluNo ratings yet
- Mathematical Modeling of Fixed Bed AdsorDocument9 pagesMathematical Modeling of Fixed Bed AdsorAl-somuda' Ali Al-smmaniNo ratings yet
- Synthesis, Characterization and Photocatalytic Activity of MnO2 Al2O3 Fe2O3 Nanocomposite For Degradation of Malachite GreenDocument12 pagesSynthesis, Characterization and Photocatalytic Activity of MnO2 Al2O3 Fe2O3 Nanocomposite For Degradation of Malachite Greenaidah amirNo ratings yet