You might also like
- BBPP 18 Colaboracion Farmaceutico Otros Profesionales SaludDocument12 pagesBBPP 18 Colaboracion Farmaceutico Otros Profesionales SaludNills ContrerasNo ratings yet
- RM 865-2019 SA Modifican-Disposiciones-Complementarias-Transitorias-Del SISMEDDocument2 pagesRM 865-2019 SA Modifican-Disposiciones-Complementarias-Transitorias-Del SISMEDNills ContrerasNo ratings yet
- Resolución Ministerial #688-2020-MINSA Directiva Administ 294 Tratamiento Datos PersonalesDocument30 pagesResolución Ministerial #688-2020-MINSA Directiva Administ 294 Tratamiento Datos PersonalesNills ContrerasNo ratings yet
- Plan de Acciones de Promocion de La Salud y Prevencion de La Enfermedad - C OvidDocument103 pagesPlan de Acciones de Promocion de La Salud y Prevencion de La Enfermedad - C OvidNills ContrerasNo ratings yet
- Plan Comunicacion 2021Document28 pagesPlan Comunicacion 2021Nills ContrerasNo ratings yet
- 03 Charlemos de Noviazgo - Es La PersonaDocument2 pages03 Charlemos de Noviazgo - Es La PersonaNills ContrerasNo ratings yet
- PERFIL DEL PROYECTO DE TESIS O INVESTIGACIÓN. DR Jhon Yagua Briceño - Epidemiologia 2017Document3 pagesPERFIL DEL PROYECTO DE TESIS O INVESTIGACIÓN. DR Jhon Yagua Briceño - Epidemiologia 2017Nills ContrerasNo ratings yet
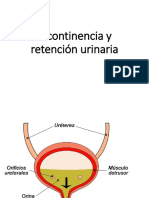
- Incontinencia y Retención UrinariaDocument38 pagesIncontinencia y Retención UrinariaNills ContrerasNo ratings yet
- Rams Causalidad.Document12 pagesRams Causalidad.Nills ContrerasNo ratings yet
- Itinerario FarmaciaDocument2 pagesItinerario FarmaciaNills ContrerasNo ratings yet
- Saint Seiya - Tomo 28 - Absorbiendo MangasDocument95 pagesSaint Seiya - Tomo 28 - Absorbiendo MangasNills ContrerasNo ratings yet
- Principios de Botanica SistematicaDocument10 pagesPrincipios de Botanica SistematicaNills Contreras100% (1)
- 001 Uso Racional de Medicamentos - CqfaDocument44 pages001 Uso Racional de Medicamentos - CqfaNills ContrerasNo ratings yet
- Noruega ResidenciaDocument4 pagesNoruega ResidenciaArbues Santa CruzNo ratings yet
- Plantilla FPI - 2 NIVEL - BucaramangaDocument6 pagesPlantilla FPI - 2 NIVEL - BucaramangaYASLEYDI TATIANA ASCANIO SANTOSNo ratings yet
- APUNTES de PAEProblemas InterdependientesDocument6 pagesAPUNTES de PAEProblemas InterdependientesteresaNo ratings yet
- Diclofenac oDocument6 pagesDiclofenac oNut Angela MarianaNo ratings yet
- Enfermedad de GoodpastureDocument1 pageEnfermedad de Goodpasturejuan7carlos7lamasNo ratings yet
- Clase 5 - Corteza Visual y Procesamiento de Imagen - TM JGBDocument41 pagesClase 5 - Corteza Visual y Procesamiento de Imagen - TM JGBJulio Cesar Barrera ReyesNo ratings yet
- Taller de Actos y Condiciones ANTAMINADocument13 pagesTaller de Actos y Condiciones ANTAMINARamon CatNo ratings yet
- Colocación de Sonda NasogastricaDocument4 pagesColocación de Sonda NasogastricaBethy VelazqueNo ratings yet
- Intervención Clínica y Psicosocial en El AncianoDocument15 pagesIntervención Clínica y Psicosocial en El Ancianorgaliano.rgNo ratings yet
- Tratamiento Homeopatico de Las Afecciones y Enfermedades Agudas - Bernardo VijnovskyDocument112 pagesTratamiento Homeopatico de Las Afecciones y Enfermedades Agudas - Bernardo VijnovskySilvana Cottié92% (12)
- AhorcamientoDocument12 pagesAhorcamientoJessica Carolay Honores ZuloagaNo ratings yet
- Espermatologia ForenseDocument25 pagesEspermatologia ForenseSATUPOPNo ratings yet
- Infeccion Vaginal en ObstetriciaDocument10 pagesInfeccion Vaginal en ObstetriciaJonatan AveigaNo ratings yet
- Guia Explicativa de Alimentación SaludableDocument24 pagesGuia Explicativa de Alimentación SaludablevivianaNo ratings yet
- Catalogo Servicios Asistenciales PDFDocument155 pagesCatalogo Servicios Asistenciales PDFunamiajaNo ratings yet
- Grupo Sanguineo 1Document25 pagesGrupo Sanguineo 1JoseloDuranNo ratings yet
- CineantropometriaDocument5 pagesCineantropometriaLILU100% (2)
- Caso Clínico: IntroducciónDocument4 pagesCaso Clínico: IntroducciónNatalia Betancur SuescunNo ratings yet
- Comparacion de Ventosas y AdhesivosDocument4 pagesComparacion de Ventosas y Adhesivosartur90998No ratings yet
- Tumores Benignos y Malignos de EsofagoDocument47 pagesTumores Benignos y Malignos de EsofagoCogake ValentinoNo ratings yet
- Glosario SNCDocument4 pagesGlosario SNCMelissa VillanuevaNo ratings yet
- Estudios ToxicológicosDocument17 pagesEstudios ToxicológicosleoNo ratings yet
- Mermelada NoniDocument5 pagesMermelada NoniluzNo ratings yet
- 2 - EstrabismoDocument39 pages2 - EstrabismoFlor GonzalesNo ratings yet
- Terapia Ocupacional y SidaDocument33 pagesTerapia Ocupacional y SidaAdalí Del Río Bermúdez0% (1)
- 3.0 Apu Covi-19 Garcia CalderonDocument4 pages3.0 Apu Covi-19 Garcia CalderonVanessa Valdivia0% (1)
- Ba Gang 8 Principios TCMDocument94 pagesBa Gang 8 Principios TCMgigicarvajalNo ratings yet
- Bioquimica Del CancerDocument31 pagesBioquimica Del Cancerjuse1213No ratings yet
- F T - Sinvirax-Oral-V0Document3 pagesF T - Sinvirax-Oral-V0dan24989No ratings yet
- CUNDUACÁNDocument30 pagesCUNDUACÁNDarwin Hernández AriasNo ratings yet