You might also like
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5795)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- (Ih Lec) 1ST ShiftDocument73 pages(Ih Lec) 1ST ShiftDennisse San JoseNo ratings yet
- Hema I Chapter 2 - Composition, Formation & Function-1Document119 pagesHema I Chapter 2 - Composition, Formation & Function-1mulugeta fentaNo ratings yet
- The Problem of Anabiosis or Latent LifeDocument44 pagesThe Problem of Anabiosis or Latent LifeAlexander ChurchillNo ratings yet
- NCP Sicu!Document6 pagesNCP Sicu!joanne190No ratings yet
- Juvenile HormoneDocument17 pagesJuvenile HormonejugesmangangNo ratings yet
- Chap17-Program Design For Resistance TrainingDocument46 pagesChap17-Program Design For Resistance TrainingPaulo CameloNo ratings yet
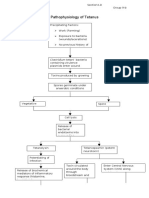
- Pathophysiology of Tetanus: Clostridium Tetani BacteriaDocument3 pagesPathophysiology of Tetanus: Clostridium Tetani BacteriaSlepy chng100% (1)
- A Case of Spinal Cord InjuryDocument2 pagesA Case of Spinal Cord InjuryliziscoolNo ratings yet
- Charge AssociationDocument2 pagesCharge AssociationBlueash BehNo ratings yet
- Active Principle: Lecture 10 Pharm Sci/Chem 177, University of California, IrvineDocument52 pagesActive Principle: Lecture 10 Pharm Sci/Chem 177, University of California, IrvineVivian PhamNo ratings yet
- Respiratory Quiz: Student Academic Learning ServicesDocument4 pagesRespiratory Quiz: Student Academic Learning ServicesFREDRICKNo ratings yet
- Lecture 3 Cell Cycle - QuestionsDocument6 pagesLecture 3 Cell Cycle - Questions中华雅思王No ratings yet
- Lambou Delto PectoralDocument9 pagesLambou Delto PectoralalexNo ratings yet
- Urinary SystemDocument31 pagesUrinary SystemAyro Business CenterNo ratings yet
- NullDocument6 pagesNullapi-24723002No ratings yet
- NAFLD - NASH and Present & Future Management OptionsDocument78 pagesNAFLD - NASH and Present & Future Management OptionsSantosh AnandNo ratings yet
- Curricullum - Health Assessment Syllabus-1Document4 pagesCurricullum - Health Assessment Syllabus-1KATZ2014No ratings yet
- Rev Notes Ch06 eDocument9 pagesRev Notes Ch06 eCHIU KEUNG OFFICIAL PRONo ratings yet
- IADVL Color Atlas of Dermatopathology - BookDocument41 pagesIADVL Color Atlas of Dermatopathology - Book65gkenNo ratings yet
- Left To Right ShuntDocument7 pagesLeft To Right Shuntanindhita anestyaNo ratings yet
- Biology - Knee Jerk SPMDocument4 pagesBiology - Knee Jerk SPMKumar AyavooNo ratings yet
- Anchorage Control in Bioprogressive Vs Straight-Wire Treatment PDFDocument6 pagesAnchorage Control in Bioprogressive Vs Straight-Wire Treatment PDFsolodont1No ratings yet
- Mls Study Guidelines AscpDocument5 pagesMls Study Guidelines AscpCielamae Orbeta100% (1)
- 1-Deep Breathing and Cough Exercises - DosdosDocument6 pages1-Deep Breathing and Cough Exercises - DosdosBianca Mikaela DosdosNo ratings yet
- ANP2001 Week 2 TutorialDocument5 pagesANP2001 Week 2 TutorialJerilee SoCute WattsNo ratings yet
- Big Picture On The Cell PosterDocument1 pageBig Picture On The Cell PosterWellcome Trust100% (1)
- Poor Diet, Lack of Sunshine and Spiritual AnemiaDocument7 pagesPoor Diet, Lack of Sunshine and Spiritual AnemiaAndri Ferdian100% (1)
- P90x-Insanity-Hybrid Workout SheetDocument1 pageP90x-Insanity-Hybrid Workout SheetMartina MartinaNo ratings yet
- Introduction To Dentistry 1Document34 pagesIntroduction To Dentistry 1Pachchi Varan100% (1)
- June 2023 (v1) (9-1) QP - Paper 4 CAIE Biology IGCSEDocument20 pagesJune 2023 (v1) (9-1) QP - Paper 4 CAIE Biology IGCSEismgilovamunisa150No ratings yet