You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Methods For Plastic RecyclingDocument19 pagesMethods For Plastic RecyclingAshik Shah100% (2)
- DHDSDocument36 pagesDHDSravichandraNo ratings yet
- Ammonia-Haldor Topsoe PDFDocument6 pagesAmmonia-Haldor Topsoe PDFShubham BansalNo ratings yet
- Chemical Recycling of PET by Catalyzed Glycolysis Kinetics of THDocument10 pagesChemical Recycling of PET by Catalyzed Glycolysis Kinetics of THViviana TeodoraNo ratings yet
- Developing A Monitoring Method Facilitating Continual Improvements in The Sorting of Waste at Recycling CentresDocument9 pagesDeveloping A Monitoring Method Facilitating Continual Improvements in The Sorting of Waste at Recycling CentresViviana TeodoraNo ratings yet
- State of The Art of Plastic Sorting and RecyclingDocument11 pagesState of The Art of Plastic Sorting and RecyclingViviana TeodoraNo ratings yet
- The Use of Electrostatic Techniques For TheDocument8 pagesThe Use of Electrostatic Techniques For TheViviana TeodoraNo ratings yet
- Zeolite WaterDocument14 pagesZeolite WaterViviana TeodoraNo ratings yet
- Electronic Waste Recyclinga Review of U.S. Infrastructure and Technology OptionsDocument33 pagesElectronic Waste Recyclinga Review of U.S. Infrastructure and Technology OptionsYesenia Najarro VarelaNo ratings yet
- Lightwight ConcreteDocument10 pagesLightwight ConcreteViviana TeodoraNo ratings yet
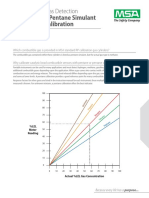
- DocumentMSA Portable Gas Detection Methane As A Pentane Simulant vs. Pentane CalibrationDocument2 pagesDocumentMSA Portable Gas Detection Methane As A Pentane Simulant vs. Pentane CalibrationgermanNo ratings yet
- Mironenko 2018Document23 pagesMironenko 2018Lê Đức HuyNo ratings yet
- 3-Enzymes (2003 - 2010)Document11 pages3-Enzymes (2003 - 2010)Asegedech BirhanuNo ratings yet
- Hydroxyapatite Synthesis and Catalytic ApplicationsDocument29 pagesHydroxyapatite Synthesis and Catalytic ApplicationsKingGeorgeVIINo ratings yet
- Singapore Chemistry Exam Guide 2017Document45 pagesSingapore Chemistry Exam Guide 2017Ng Shu QingNo ratings yet
- DTE Energy/EES Coke PTI 51-08C FactSheetDocument16 pagesDTE Energy/EES Coke PTI 51-08C FactSheetStephen BoyleNo ratings yet
- SAT Chemistry Eng-11-12 G AdvancedDocument80 pagesSAT Chemistry Eng-11-12 G AdvancedАрнур ОспановNo ratings yet
- PPTDocument17 pagesPPTArvind SinghNo ratings yet
- FCH - ChE 331A - JAN - 2019Document2 pagesFCH - ChE 331A - JAN - 2019Abhinav ShuklaNo ratings yet
- Transport Processes and Carrier DesignDocument24 pagesTransport Processes and Carrier Designcgckamilshaikh50% (2)
- مصدر المطلوبDocument12 pagesمصدر المطلوبaliNo ratings yet
- Carbon Deposition in Heterogeneous Catalysis: Solved With Comsol Reaction Engineering Lab 3.5ADocument13 pagesCarbon Deposition in Heterogeneous Catalysis: Solved With Comsol Reaction Engineering Lab 3.5AStephanie WalkerNo ratings yet
- Dispersion For CeramicDocument5 pagesDispersion For CeramicThanhNo ratings yet
- 414 - LONG TEST 1 To 2Document14 pages414 - LONG TEST 1 To 2CapsanneNo ratings yet
- First Row Transition MetalsDocument39 pagesFirst Row Transition MetalsArielle LewisNo ratings yet
- Us 6677477Document8 pagesUs 6677477nurhafizah jabarNo ratings yet
- Enzyme Assays Based On UV-VIS SpectrophotometerDocument12 pagesEnzyme Assays Based On UV-VIS SpectrophotometerYusof SundangNo ratings yet
- Hydrogen Evolution Reaction On NiCu Electrodeposited Electrodes in 6.0 M KOHDocument5 pagesHydrogen Evolution Reaction On NiCu Electrodeposited Electrodes in 6.0 M KOHVỹ UôngNo ratings yet
- Spectrum Chemistry - May 2016Document100 pagesSpectrum Chemistry - May 2016Shailesh prajapatiNo ratings yet
- Oxidation of Alcohols With Hydrogen Peroxide in The Presence of A New Triple-Site PhosphotungstateDocument11 pagesOxidation of Alcohols With Hydrogen Peroxide in The Presence of A New Triple-Site PhosphotungstatexcvNo ratings yet
- Enzymes AssignmentDocument2 pagesEnzymes AssignmentBibek GajmerNo ratings yet
- Instruction Manual TA2000Document35 pagesInstruction Manual TA2000Roshin99No ratings yet
- 172 182 JMTR Jul17Document11 pages172 182 JMTR Jul17Mikel MichaelNo ratings yet
- RP15C-toc 173390110917062932Document13 pagesRP15C-toc 173390110917062932manav mistryNo ratings yet
- Principles of Heterogeneous CatalysisDocument15 pagesPrinciples of Heterogeneous CatalysisHector Martinez HernandezNo ratings yet
- 2016-Small - Graphene in Photocatalysis A Review PDFDocument57 pages2016-Small - Graphene in Photocatalysis A Review PDFsneha mallikaNo ratings yet
- Methanol To Olefins (MTO) : Development of A Commercial Catalytic ProcessDocument98 pagesMethanol To Olefins (MTO) : Development of A Commercial Catalytic ProcessРоман ЗахаровNo ratings yet
- Programme FINALDocument27 pagesProgramme FINALRafael BarrosNo ratings yet