You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- KetamineDocument23 pagesKetamineDimas Gatra DiantoroNo ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Your Body Believes Every Word Y - Barbara Hoberman LevineDocument380 pagesYour Body Believes Every Word Y - Barbara Hoberman LevineRoryBradshawNo ratings yet
- Borderline Personality Disorder BSN 3y2-3a G3Document24 pagesBorderline Personality Disorder BSN 3y2-3a G3SHARMAINE ANNE POLICIOS100% (2)
- A Lead CompoundDocument2 pagesA Lead CompoundfikrifazNo ratings yet
- Fundamentals of NursingDocument50 pagesFundamentals of NursingGuruKPO67% (3)
- Connections, Displays and Operating Elements: MTN299200/MTN299201/MTN299202Document2 pagesConnections, Displays and Operating Elements: MTN299200/MTN299201/MTN299202fikrifazNo ratings yet
- Stephen Danso - MensahDocument126 pagesStephen Danso - MensahfikrifazNo ratings yet
- WHO MONO 27 (2ed) (chp6)Document32 pagesWHO MONO 27 (2ed) (chp6)fikrifazNo ratings yet
- WHO MONO 27 (2ed) (chp6)Document32 pagesWHO MONO 27 (2ed) (chp6)fikrifazNo ratings yet
- Prop y Lene GlycolDocument2 pagesProp y Lene GlycolfikrifazNo ratings yet
- Fullprof IntroDocument14 pagesFullprof IntrofikrifazNo ratings yet
- Abstracted Indexed JocprDocument7 pagesAbstracted Indexed JocprfikrifazNo ratings yet
- Torpac Rat Gpig PricingDocument1 pageTorpac Rat Gpig PricingfikrifazNo ratings yet
- Additive 369Document3 pagesAdditive 369fikrifazNo ratings yet
- Shimadzu FTIR Academic 12-1Document1 pageShimadzu FTIR Academic 12-1fikrifazNo ratings yet
- TB Solubility SaccharinDocument1 pageTB Solubility SaccharinfikrifazNo ratings yet
- TB Solubility SaccharinDocument1 pageTB Solubility SaccharinfikrifazNo ratings yet
- Torpac Rat Gpig PricingDocument1 pageTorpac Rat Gpig PricingfikrifazNo ratings yet
- Injeksi SolvochinDocument1 pageInjeksi SolvochinfikrifazNo ratings yet
- Shimadzu FTIR Academic 12-1Document1 pageShimadzu FTIR Academic 12-1fikrifazNo ratings yet
- Torpac Rat Gpig PricingDocument1 pageTorpac Rat Gpig PricingfikrifazNo ratings yet
- Prop y Lene GlycolDocument2 pagesProp y Lene GlycolfikrifazNo ratings yet
- Torpac Rat Gpig PricingDocument1 pageTorpac Rat Gpig PricingfikrifazNo ratings yet
- Prop y Lene GlycolDocument2 pagesProp y Lene GlycolfikrifazNo ratings yet
- tp189 c6Document7 pagestp189 c6fikrifazNo ratings yet
- Torpac Rat Gpig PricingDocument1 pageTorpac Rat Gpig PricingfikrifazNo ratings yet
- Quantitative and Qualitative Analysis of Medication Errors: The New York ExperienceDocument14 pagesQuantitative and Qualitative Analysis of Medication Errors: The New York ExperiencefikrifazNo ratings yet
- IR Absorption TableDocument2 pagesIR Absorption TablefikrifazNo ratings yet
- 1713 BP 1Document17 pages1713 BP 1api-3698598No ratings yet
- Medical Books December 2010Document21 pagesMedical Books December 2010matchealthNo ratings yet
- Blood Transfusion Reactions: P Sunil Kumar Department of Haematology ST - John's Medical CollegeDocument48 pagesBlood Transfusion Reactions: P Sunil Kumar Department of Haematology ST - John's Medical CollegeFULGENCE RUHARARANo ratings yet
- Spine Magnetic Resonance Image Segmentation Using Deep Learning TechniquesDocument6 pagesSpine Magnetic Resonance Image Segmentation Using Deep Learning TechniquesSandeep VermaNo ratings yet
- Internet Addiction Kids and The South Korean Govt Boot CampsDocument5 pagesInternet Addiction Kids and The South Korean Govt Boot Campslgheorma0No ratings yet
- Struggle and Survival of Native Americans: A Study in Selected Poems by Simon J. OrtizDocument15 pagesStruggle and Survival of Native Americans: A Study in Selected Poems by Simon J. OrtizCycilian ArmandoNo ratings yet
- Asthma and Diabetes P1 P2 P3Document6 pagesAsthma and Diabetes P1 P2 P3desbestNo ratings yet
- 1116005I Rev. 02Document2 pages1116005I Rev. 02kirubel demelashNo ratings yet
- HePatic AbscessDocument67 pagesHePatic AbscessCharlie Mignonette BalaNo ratings yet
- Nuggets Combined - Indexed + Searchable PDFDocument478 pagesNuggets Combined - Indexed + Searchable PDFjasleenNo ratings yet
- Log RegDocument32 pagesLog RegSIDDHARTH KUMARNo ratings yet
- 17-10 eJDD FinalDocument116 pages17-10 eJDD FinalMohamed GamalNo ratings yet
- Giant Cell TumourDocument61 pagesGiant Cell Tumourvictormoirangthem100% (1)
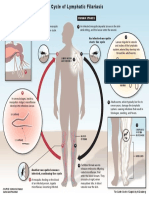
- Lymphatic Filariasis Life Cycle PDFDocument1 pageLymphatic Filariasis Life Cycle PDFlauraNo ratings yet
- Neurotrac Tens: Operators ManualDocument28 pagesNeurotrac Tens: Operators ManualShane NaidooNo ratings yet
- Bivalirudin Anticoagulant1Document6 pagesBivalirudin Anticoagulant1walid hassanNo ratings yet
- EMCrit Lae Pulmonary FlowDocument1 pageEMCrit Lae Pulmonary FlowhmsptrNo ratings yet
- Chapter 39: Plant Responses To Internal and External SignalsDocument9 pagesChapter 39: Plant Responses To Internal and External SignalsdarthNo ratings yet
- Mosquito Net SpecificationDocument12 pagesMosquito Net SpecificationManoj KumarNo ratings yet
- Human Anatomy & Physiology: Chapter 21-1Document103 pagesHuman Anatomy & Physiology: Chapter 21-1AngelyNo ratings yet
- A Review Paper On Scope of Big Data Analysis in Heath INFORMATICSDocument8 pagesA Review Paper On Scope of Big Data Analysis in Heath INFORMATICSMohamed Aly SowNo ratings yet
- Chapter 4 - Ornamental FishDocument34 pagesChapter 4 - Ornamental FishJuan Carlos SánchezNo ratings yet
- Status of Biological Control in MealybugDocument19 pagesStatus of Biological Control in MealybugDinesh KumarNo ratings yet
- ZOOLOGY LESSON 1 IntroductionbranchesofzoologyDocument25 pagesZOOLOGY LESSON 1 IntroductionbranchesofzoologyLeimarie BarsanaNo ratings yet
- ENT Atlas Web PDFDocument442 pagesENT Atlas Web PDFIsabella CrăciunNo ratings yet
- Lab 1C PAR-Q and Health History QuestionnaireDocument4 pagesLab 1C PAR-Q and Health History Questionnaireskyward23No ratings yet
- Uji Beberapa Konsentrasi Cendawan EntomoDocument16 pagesUji Beberapa Konsentrasi Cendawan EntomoAngger DimasNo ratings yet