You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Pharmacology Reasons and Effect On SocietyDocument7 pagesPharmacology Reasons and Effect On SocietyAlejandro Arévalo RamosNo ratings yet
- 1 1 6 Final DiagnosisDocument1 page1 1 6 Final Diagnosisapi-261996621No ratings yet
- Swiss Guidelines For Safe 1.7 Dry Needling 01Document21 pagesSwiss Guidelines For Safe 1.7 Dry Needling 01monkeyhsuNo ratings yet
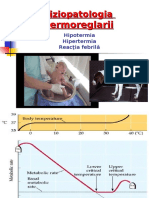
- Documents - MX Fiziopatologia Termoreglarii Fiziopatologia Termoreglarii Hipotermia Hipertermia Reactia Febrila 10-12-2006Document42 pagesDocuments - MX Fiziopatologia Termoreglarii Fiziopatologia Termoreglarii Hipotermia Hipertermia Reactia Febrila 10-12-2006AndreiNo ratings yet
- 1.1 Role of Organic Drug Molecules Unit 1Document16 pages1.1 Role of Organic Drug Molecules Unit 1Amarnath SahNo ratings yet
- CPR Ratio For Adults, Children, and InfantsDocument14 pagesCPR Ratio For Adults, Children, and InfantsAnonymous FCOOcnNo ratings yet
- Sterilization of DogsDocument18 pagesSterilization of Dogsjo12joseNo ratings yet
- Perioperative Fluid ManagementDocument123 pagesPerioperative Fluid ManagementAnonymous 86gki5No ratings yet
- BeyondChemo WorkBookDocument42 pagesBeyondChemo WorkBookgreentree14No ratings yet
- Research Into Complementary and Alternative Medicine: Problems and PotentialDocument4 pagesResearch Into Complementary and Alternative Medicine: Problems and PotentialCyntia GavrilaNo ratings yet
- Im Residency FlowchartDocument1 pageIm Residency FlowchartF Badruzzama BegumNo ratings yet
- What Is Biliary Atresia?Document6 pagesWhat Is Biliary Atresia?echi_maniezz8719No ratings yet
- Stem Cell Therapy For Rotator Cuff InjuriesDocument12 pagesStem Cell Therapy For Rotator Cuff InjuriesAthenaeum Scientific PublishersNo ratings yet
- NCM 109 Reviewer Module 1Document6 pagesNCM 109 Reviewer Module 1tamsmadjad18No ratings yet
- Anaesthetic Case StudyDocument11 pagesAnaesthetic Case StudysomyntNo ratings yet
- A Love Affair That Got Me Close To A Great DoctorDocument2 pagesA Love Affair That Got Me Close To A Great DoctorRudolf Dwight MaqueraNo ratings yet
- UWorld Notes Step 2 CKDocument11 pagesUWorld Notes Step 2 CKSamah Khan50% (2)
- Contemporary Implant Dentistry: Chapter 14 of Contemporary Oral and Maxillofacial Surgery By: DrarashkhojastehDocument65 pagesContemporary Implant Dentistry: Chapter 14 of Contemporary Oral and Maxillofacial Surgery By: DrarashkhojastehRawa Omar100% (1)
- DQoL Jacobson 1988 PDFDocument8 pagesDQoL Jacobson 1988 PDFeunikieNo ratings yet
- Ovarian Cancer: A. IntroductionDocument10 pagesOvarian Cancer: A. IntroductionBer AnneNo ratings yet
- 23-Discharge Against Medical Advice (Dama)Document3 pages23-Discharge Against Medical Advice (Dama)akositabonNo ratings yet
- Label Nama Obat Di Rak ObatDocument6 pagesLabel Nama Obat Di Rak Obatanon_756054910No ratings yet
- Effectiveness_of_Hydrotherapy_ (1)Document18 pagesEffectiveness_of_Hydrotherapy_ (1)dandiokasubantaraNo ratings yet
- 491-Article Text-2225-1-10-20230328Document7 pages491-Article Text-2225-1-10-20230328Atik TriyanaNo ratings yet
- Jurding Otosklerosis Nor Azmina 112017074Document19 pagesJurding Otosklerosis Nor Azmina 112017074beatriceNo ratings yet
- Oxygen TherapyDocument20 pagesOxygen TherapyPaul Gabriel CasquejoNo ratings yet
- 77.18 Shen Guan and Three Emperors - Master Kidney PointDocument3 pages77.18 Shen Guan and Three Emperors - Master Kidney Pointalbar ismuNo ratings yet
- PHYSIOLOGICAL EFFECTS OF VALSALVA AND IPPVDocument27 pagesPHYSIOLOGICAL EFFECTS OF VALSALVA AND IPPVChaturika Bandara100% (1)
- Introduction To JubileeDocument117 pagesIntroduction To Jubileeangela gnanaduraiNo ratings yet
- Uterine ProlapseDocument3 pagesUterine ProlapseKeshavprasad kurmi100% (1)