You might also like
- Tian 2011 Vacuum 1Document7 pagesTian 2011 Vacuum 1momenziNo ratings yet
- 076 PDFDocument6 pages076 PDFprakush01975225403No ratings yet
- TMP 47 A7Document13 pagesTMP 47 A7FrontiersNo ratings yet
- Electrochemical Behavior of Layered Solid Solution Li Mno 2limo (M 5 Ni, MN, Co) Li-Ion Cathodes With and Without Alumina CoatingsDocument7 pagesElectrochemical Behavior of Layered Solid Solution Li Mno 2limo (M 5 Ni, MN, Co) Li-Ion Cathodes With and Without Alumina CoatingsEYERUSALEM TADESSENo ratings yet
- Electrodeposition of Zinc-Tin Alloys From Deep Eutectic Solvents Based On Choline ChlorideDocument7 pagesElectrodeposition of Zinc-Tin Alloys From Deep Eutectic Solvents Based On Choline ChlorideJohnSmithNo ratings yet
- Isotherm 1Document11 pagesIsotherm 1HemrajNo ratings yet
- Taylor 2017Document12 pagesTaylor 2017alvaro rodriguez molina do santosNo ratings yet
- Electrolyzer Modules A) Unipolar and B) Bipolar Cell ConfigurationsDocument8 pagesElectrolyzer Modules A) Unipolar and B) Bipolar Cell ConfigurationsAnuj ShahiNo ratings yet
- Study of Corrosion Inhibition of Mild Steel in Acidic Medium by 2-Propargyl-5-P-chlorophenyltetrazole - Part IDocument8 pagesStudy of Corrosion Inhibition of Mild Steel in Acidic Medium by 2-Propargyl-5-P-chlorophenyltetrazole - Part ILWYenNo ratings yet
- Treatment of Wastewater by Electro Coagulation: A Review: ISO 9001:2008 CertifiedDocument7 pagesTreatment of Wastewater by Electro Coagulation: A Review: ISO 9001:2008 CertifiedImane BendarouachNo ratings yet
- AC DC Studies On CI of Low Carbon Steel in HCL by Succinic Acid 2008Document17 pagesAC DC Studies On CI of Low Carbon Steel in HCL by Succinic Acid 2008gabriel norbertNo ratings yet
- Cyclic Voltammetry and Scanning Electrochemical Microscopy of FerrocementhanolDocument9 pagesCyclic Voltammetry and Scanning Electrochemical Microscopy of Ferrocementhanolhongluc1991No ratings yet
- J Cattod 2011 07 008Document8 pagesJ Cattod 2011 07 008Syeda Ammara AnwarNo ratings yet
- A Quantitative Comparison Between Chemical Dosing and ElectrocoagulationDocument16 pagesA Quantitative Comparison Between Chemical Dosing and Electrocoagulationlusi.meliyanaNo ratings yet
- Electrowinning of Cobalt From Acidic Sulphate Solutions-Effect of Chloride IonDocument9 pagesElectrowinning of Cobalt From Acidic Sulphate Solutions-Effect of Chloride IontabatabayiNo ratings yet
- Electrochemical Degradability of Al-20% MG and Al-22% Si Alloys in An Acidic Environment in Relation With Their MicrostructureDocument20 pagesElectrochemical Degradability of Al-20% MG and Al-22% Si Alloys in An Acidic Environment in Relation With Their MicrostructureAishah SamNo ratings yet
- Biolix Calco A Distintas Temp.Document14 pagesBiolix Calco A Distintas Temp.Feña Aranda DelaFuenteNo ratings yet
- Study of The Underlying Electrochemistry of Polycrystalline GoldDocument13 pagesStudy of The Underlying Electrochemistry of Polycrystalline GoldAzucena osornio villaNo ratings yet
- 1 s2.0 S1452398123152849 MainDocument17 pages1 s2.0 S1452398123152849 MainKarolina GawlakNo ratings yet
- PH.D - Synopsis - M. Praveen KumarDocument12 pagesPH.D - Synopsis - M. Praveen KumaralexabcdxyzNo ratings yet
- Electrodeposition of Copper From Non-Cyanide AlkalineDocument20 pagesElectrodeposition of Copper From Non-Cyanide AlkalineDerdo ZulmuNo ratings yet
- A Carbon Electrode Fabricated Using A Poly (Vinylidene Fluoride) Binder Controlled The Faradaic Reaction of Carbon PowderDocument5 pagesA Carbon Electrode Fabricated Using A Poly (Vinylidene Fluoride) Binder Controlled The Faradaic Reaction of Carbon PowderAlice1923No ratings yet
- Energies 05 05363 v2Document9 pagesEnergies 05 05363 v2mevlut46No ratings yet
- I Nfluence of Pore Structure and Surface Chemistry On Electric Double Layer Capacitance in Non-Aqueous ElectrolyteDocument11 pagesI Nfluence of Pore Structure and Surface Chemistry On Electric Double Layer Capacitance in Non-Aqueous ElectrolyteJerusa Pacheco SampaioNo ratings yet
- Abraham, Yusuff - 2003 - Copper (II) Complexes of Embelin and 2-Aminobenzimidazole Encapsulated in Zeolite Y-Potential As Catalysts For RDocument9 pagesAbraham, Yusuff - 2003 - Copper (II) Complexes of Embelin and 2-Aminobenzimidazole Encapsulated in Zeolite Y-Potential As Catalysts For Rcukaasam123456No ratings yet
- Fundamentals Present and Future Perspect PDFDocument12 pagesFundamentals Present and Future Perspect PDFFebrianNo ratings yet
- Paper 6Document7 pagesPaper 6AdityaNo ratings yet
- Ion ExchangeDocument13 pagesIon Exchangebreadfalling752No ratings yet
- Electrochemical Impedance Models For Molten Salt Corrosion: C.L. Zeng, W. Wang, W.T. WuDocument15 pagesElectrochemical Impedance Models For Molten Salt Corrosion: C.L. Zeng, W. Wang, W.T. WuVictor SabNo ratings yet
- Applied Surface Science 347 (2015) 40-47Document8 pagesApplied Surface Science 347 (2015) 40-47Hugo DuarteNo ratings yet
- Electrochemical Performance of A Solvent-Free Hybrid Ceramic - Keller - J Power Sources 2017Document11 pagesElectrochemical Performance of A Solvent-Free Hybrid Ceramic - Keller - J Power Sources 2017I.T.S EnggNo ratings yet
- Electrochemistry Communications: Lin Yu, Jizhou Duan, Xiangqian Du, Yanliang Huang, Baorong HouDocument4 pagesElectrochemistry Communications: Lin Yu, Jizhou Duan, Xiangqian Du, Yanliang Huang, Baorong HouAnushri VaidyaNo ratings yet
- Lithium Air Battery ThesisDocument6 pagesLithium Air Battery Thesisalissacruzomaha100% (2)
- Electrochemistry Communications: Lin Yu, Jizhou Duan, Xiangqian Du, Yanliang Huang, Baorong HouDocument4 pagesElectrochemistry Communications: Lin Yu, Jizhou Duan, Xiangqian Du, Yanliang Huang, Baorong HouAnushri VaidyaNo ratings yet
- 25 - 2015 - Nat - Comm - Bifunctional Non-Noble Metal Oxide Nanoparticle ElectrocatalystsDocument9 pages25 - 2015 - Nat - Comm - Bifunctional Non-Noble Metal Oxide Nanoparticle ElectrocatalystsCB Dong SuwonNo ratings yet
- Roto 2002Document8 pagesRoto 2002k.suganeswaranNo ratings yet
- Waste Water ManagementDocument11 pagesWaste Water ManagementViknesh VikiNo ratings yet
- TMP 2 B55Document24 pagesTMP 2 B55FrontiersNo ratings yet
- Mass Loading Optimization For Ethylene Glycol Oxidation at Different Potential RegionsDocument9 pagesMass Loading Optimization For Ethylene Glycol Oxidation at Different Potential RegionsCB Dong SuwonNo ratings yet
- Hydrogen Evolution Reaction On NiCu Electrodeposited Electrodes in 6.0 M KOHDocument5 pagesHydrogen Evolution Reaction On NiCu Electrodeposited Electrodes in 6.0 M KOHVỹ UôngNo ratings yet
- Glassy Carbon ElectrodesDocument11 pagesGlassy Carbon ElectrodesTiana JovanovicNo ratings yet
- 1 s2.0 S0167273822000376 MainDocument7 pages1 s2.0 S0167273822000376 MainAkrithi AkrithiNo ratings yet
- Direct Electrochemical Reduction of Indigo: Process Optimization and Scale-Up in A Ow CellDocument5 pagesDirect Electrochemical Reduction of Indigo: Process Optimization and Scale-Up in A Ow CellKarrar HaiderNo ratings yet
- Resnik MEET2019Document7 pagesResnik MEET2019Surya Chandra NamahaNo ratings yet
- Anodic Reactions: 08Y506 - Corrosion EngineeringDocument5 pagesAnodic Reactions: 08Y506 - Corrosion Engineeringesakkibabu1987No ratings yet
- Graphene Oxide Modified Lini Co MN O As Cathode Material For Lithium Ion Batteries and Its Electrochemical PerformancesDocument14 pagesGraphene Oxide Modified Lini Co MN O As Cathode Material For Lithium Ion Batteries and Its Electrochemical Performancesआदित्य नारायण सिंहNo ratings yet
- Study of The Electrocoagulation of Electroplating Industry Wastewaters Charged by Nickel (II) and Chromium (VI)Document10 pagesStudy of The Electrocoagulation of Electroplating Industry Wastewaters Charged by Nickel (II) and Chromium (VI)Martin FernandezNo ratings yet
- 1 s2.0 S0013468613011997 MainDocument7 pages1 s2.0 S0013468613011997 MainDang MinhNo ratings yet
- Carbon Film Electrodes As Support of Metallic Particles: Int. J. Electrochem. Sci., 7 (2012) 150 - 166Document17 pagesCarbon Film Electrodes As Support of Metallic Particles: Int. J. Electrochem. Sci., 7 (2012) 150 - 166FelpnilNo ratings yet
- Assignment 2Document4 pagesAssignment 2tinashe tagariraNo ratings yet
- Antimicrobial and Anticorrosive Activity of Adsorbents Based On Chitosan Schiff's BaseDocument21 pagesAntimicrobial and Anticorrosive Activity of Adsorbents Based On Chitosan Schiff's BaseFernanda Stuani PereiraNo ratings yet
- Facile Synthesis of CuO - ITO Film Via The Chronoamperometric Electrodeposition For Nonenzymatic Glucose SensingDocument10 pagesFacile Synthesis of CuO - ITO Film Via The Chronoamperometric Electrodeposition For Nonenzymatic Glucose SensingDũng Quốc NguyễnNo ratings yet
- A Systematic Study On Electrolytic Production of Hydrogen Gas by Using Graphite As ElectrodeDocument5 pagesA Systematic Study On Electrolytic Production of Hydrogen Gas by Using Graphite As ElectrodeZahra AlifiaNo ratings yet
- GravimetricDocument20 pagesGravimetricHaniel FcNo ratings yet
- Materials LettersDocument4 pagesMaterials LettersSahin CoskunNo ratings yet
- Corrosion Inhibition of Copper in NitricDocument17 pagesCorrosion Inhibition of Copper in Nitriclorenaov177No ratings yet
- Amperometric End-Point Detection of ComplexometricDocument6 pagesAmperometric End-Point Detection of ComplexometricSteven John PadillaNo ratings yet
- Adsorption and Corrosion Inhibition of New Synthesized Pyridazinium-Based Ionic Liquid On Carbon Steel in 0.5 M H SODocument9 pagesAdsorption and Corrosion Inhibition of New Synthesized Pyridazinium-Based Ionic Liquid On Carbon Steel in 0.5 M H SOHyd BenNo ratings yet
- Biofilms in Bioelectrochemical Systems: From Laboratory Practice to Data InterpretationFrom EverandBiofilms in Bioelectrochemical Systems: From Laboratory Practice to Data InterpretationNo ratings yet
- Supercapacitors Based on Carbon or Pseudocapacitive MaterialsFrom EverandSupercapacitors Based on Carbon or Pseudocapacitive MaterialsNo ratings yet
- Selective Extraction of Neutral Nitrogen Compounds Found in Diesel Feed byDocument8 pagesSelective Extraction of Neutral Nitrogen Compounds Found in Diesel Feed byJohnSmithNo ratings yet
- Selective Adsorption For Removal of Nitrogen Compounds From Liquid HC Streams Over Carbon - and Alumina - Based AdsorbentsDocument10 pagesSelective Adsorption For Removal of Nitrogen Compounds From Liquid HC Streams Over Carbon - and Alumina - Based AdsorbentsJohnSmithNo ratings yet
- Zn-Containing Ionic Liquids For The Extractive Denitrogenation of A Model Oil - A Mechanistic ConsiderationDocument7 pagesZn-Containing Ionic Liquids For The Extractive Denitrogenation of A Model Oil - A Mechanistic ConsiderationJohnSmithNo ratings yet
- Inhibition Effects of Nitrogen Compounds On The HDS of Dibenzothiophene Part 2aDocument8 pagesInhibition Effects of Nitrogen Compounds On The HDS of Dibenzothiophene Part 2aJohnSmithNo ratings yet
- Are Deep EutecticDocument3 pagesAre Deep EutecticJohnSmithNo ratings yet
- Science:, 792 (2003) Robin D. Rogers and Kenneth R. SeddonDocument3 pagesScience:, 792 (2003) Robin D. Rogers and Kenneth R. SeddonJohnSmithNo ratings yet
- Post-Etch Residue Removal Using Choline Chloride-Malonic Acid Deep Eutectic Solvent (DES)Document6 pagesPost-Etch Residue Removal Using Choline Chloride-Malonic Acid Deep Eutectic Solvent (DES)JohnSmithNo ratings yet
- Molar Heat Capacities of Choline Chloride-Based Deep Eutectic Solvents and Their Binary Mixtures With WaterDocument25 pagesMolar Heat Capacities of Choline Chloride-Based Deep Eutectic Solvents and Their Binary Mixtures With WaterJohnSmithNo ratings yet
- A Simplified Method For The Cultivation of Extreme Anaerobic Archaea Based SULFIDE 2000 !!!!Document6 pagesA Simplified Method For The Cultivation of Extreme Anaerobic Archaea Based SULFIDE 2000 !!!!Vera Brok-VolchanskayaNo ratings yet
- Full Solution Manual For Chemistry For Changing Times 14Th Edition John W Hill Terry W Mccreary PDF Docx Full Chapter ChapterDocument34 pagesFull Solution Manual For Chemistry For Changing Times 14Th Edition John W Hill Terry W Mccreary PDF Docx Full Chapter Chapterseesaw.insearchd8k4100% (15)
- 2007 LE Mayer TwleveDocument5 pages2007 LE Mayer TwleveVictor ZhicayNo ratings yet
- Sci AvogadroDocument3 pagesSci AvogadroFiona Antoinette BesaNo ratings yet
- PH - Wikipedia, The Free EncyclopediaDocument11 pagesPH - Wikipedia, The Free EncyclopediaShikhar MahajanNo ratings yet
- BS en 10222-2 - 2017 PDFDocument32 pagesBS en 10222-2 - 2017 PDFАртем Титов75% (4)
- Chemistry Education in The 21st CenturyDocument106 pagesChemistry Education in The 21st CenturyAbhishek BansalNo ratings yet
- Chemistry Practical Exam TipsDocument12 pagesChemistry Practical Exam Tipshaya waqrNo ratings yet
- Year 9 Study Guide ChemistryDocument15 pagesYear 9 Study Guide Chemistryreem halawiNo ratings yet
- Modelling of Aluminium Scrap Melting in A Rotary FurnaceDocument10 pagesModelling of Aluminium Scrap Melting in A Rotary FurnaceestefanoveiraNo ratings yet
- Pre-IB Chemistry Mid-Term Review List (Nagel)Document3 pagesPre-IB Chemistry Mid-Term Review List (Nagel)Helie100% (1)
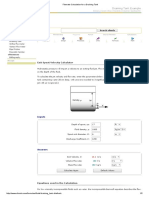
- Flowrate Calculation For A Draining TankDocument2 pagesFlowrate Calculation For A Draining TankAnonymous bHh1L1No ratings yet
- Finalreport PDFDocument47 pagesFinalreport PDFNelCamHerNo ratings yet
- StereochemistryDocument52 pagesStereochemistryTimmyNo ratings yet
- Reichold, Corrosion Guide 12.15.2010Document45 pagesReichold, Corrosion Guide 12.15.2010Pradeep Srivastava100% (1)
- LOVIBOND Reagents Suited For HachDocument6 pagesLOVIBOND Reagents Suited For HachKacem BenaoumeurNo ratings yet
- Stabilityindicating HPTLC Method For Simultaneous Estimation of Amoxicillin Trihydrate and Ambroxol Hydrochloride in Bulk and Pharmaceutical Dosage Form 2153 2435-4-261Document5 pagesStabilityindicating HPTLC Method For Simultaneous Estimation of Amoxicillin Trihydrate and Ambroxol Hydrochloride in Bulk and Pharmaceutical Dosage Form 2153 2435-4-261Fadhil Muhammad AwaluddinNo ratings yet
- CY100 Engineering Chemistry Syllabus 2016Document2 pagesCY100 Engineering Chemistry Syllabus 2016Siju N. AntonyNo ratings yet
- 2007 - Shellac in PolymerDocument43 pages2007 - Shellac in PolymerAnonymous x7VY8VF7No ratings yet
- SilaneCouplingAgents eDocument24 pagesSilaneCouplingAgents eElisabeth Kurnia Bloom100% (1)
- Lecture Notes On ThermoelectricityDocument21 pagesLecture Notes On ThermoelectricityBradley NartowtNo ratings yet
- PS1Document4 pagesPS1cptudorNo ratings yet
- Shell Gadus s1 Og 200 PDFDocument1 pageShell Gadus s1 Og 200 PDFMiskaDarainiNo ratings yet
- RioBooster SDSDocument10 pagesRioBooster SDSpepeNo ratings yet
- Microsoft Word - Slickline Mechanical CapibilitiesDocument7 pagesMicrosoft Word - Slickline Mechanical CapibilitiesRangga DraApNo ratings yet
- Full Download General Organic and Biological Chemistry 2nd Edition Janice Gorzynski Smith Test Bank PDF Full ChapterDocument36 pagesFull Download General Organic and Biological Chemistry 2nd Edition Janice Gorzynski Smith Test Bank PDF Full Chapternuggetessayistypcu100% (18)
- Chemical Reactions and Equations Assignment Questions Set - 1Document6 pagesChemical Reactions and Equations Assignment Questions Set - 1kayace8055No ratings yet
- GUI Waste Incineration & Best Available Techniques (BAT) - Bref2004Document530 pagesGUI Waste Incineration & Best Available Techniques (BAT) - Bref2004susCities100% (7)
- Stainless Steel Braided Teflon Flexible Hoses Hht&Hhts SeriesDocument4 pagesStainless Steel Braided Teflon Flexible Hoses Hht&Hhts SeriesMKOZERDEMNo ratings yet
- Oxygen Unit Calculation Dv1 1Document2 pagesOxygen Unit Calculation Dv1 1Gisele CescuttiNo ratings yet