You might also like
- 2014 - Genomic Characterization of Ependymomas RevealsDocument8 pages2014 - Genomic Characterization of Ependymomas RevealsIgor PessôaNo ratings yet
- 2012-A Huge Intraventricular Congenital Anaplastic Astrocytoma CaseDocument6 pages2012-A Huge Intraventricular Congenital Anaplastic Astrocytoma CaseIgor PessôaNo ratings yet
- Ceccarelli 2016Document15 pagesCeccarelli 2016Igor PessôaNo ratings yet
- 2005-Molecular Cytogenetic Analysis in The Study of Brain TumorsDocument36 pages2005-Molecular Cytogenetic Analysis in The Study of Brain TumorsIgor PessôaNo ratings yet
- 2011 Molecular CytogeneticsDocument15 pages2011 Molecular CytogeneticsIgor PessôaNo ratings yet
- Spirituality and Religion in OncologyDocument10 pagesSpirituality and Religion in OncologyIgor PessôaNo ratings yet
- Guia Programa de MetilaçãoDocument40 pagesGuia Programa de MetilaçãoIgor PessôaNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (120)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Material Cost - EOQDocument16 pagesMaterial Cost - EOQSimranNo ratings yet
- For Student-General Chemistry I - Module 6 - Phan Tai HuanDocument41 pagesFor Student-General Chemistry I - Module 6 - Phan Tai HuanEsat Goceri100% (1)
- 00 - Orientation LessonDocument32 pages00 - Orientation LessonRekha ShahNo ratings yet
- ASME B36.10M-2004 Welded and Seamless Wrought Steel Pipe StandardDocument2 pagesASME B36.10M-2004 Welded and Seamless Wrought Steel Pipe StandardAmit BansalNo ratings yet
- NAS810 Protection of Fluid Lines & EquipDocument1 pageNAS810 Protection of Fluid Lines & EquipfdhgjklNo ratings yet
- Meng 2nd Year Death Anniversary MassDocument6 pagesMeng 2nd Year Death Anniversary Massjoy in the spirit of the lordNo ratings yet
- Pneumatic System and Basic Valve UsedDocument401 pagesPneumatic System and Basic Valve Usedtarang srivasNo ratings yet
- Honeywell TH6210U2001 Install InstructionsDocument44 pagesHoneywell TH6210U2001 Install Instructionsdarwin jose palacioNo ratings yet
- FIITJEE SAMPLE PAPER – 2018 (Big Bang Edge Test / Talent Recognition Exam) for Class 10 (Paper 2Document17 pagesFIITJEE SAMPLE PAPER – 2018 (Big Bang Edge Test / Talent Recognition Exam) for Class 10 (Paper 2msreddy86No ratings yet
- Drug Metabolism in Fetus and NewbornsDocument20 pagesDrug Metabolism in Fetus and NewbornsBikash SahNo ratings yet
- Viral Skin Infections Caused by Herpesviridae and PoxviridaeDocument55 pagesViral Skin Infections Caused by Herpesviridae and PoxviridaeGita RizkiNo ratings yet
- Standard Operating Procedure: Title: Materials Control - Work in Process (WIP) ApprovalsDocument2 pagesStandard Operating Procedure: Title: Materials Control - Work in Process (WIP) ApprovalsRAHUL YADAV100% (1)
- Engineering Mathematics For Gate Chapter1Document1 pageEngineering Mathematics For Gate Chapter1Sai VeerendraNo ratings yet
- Prog 4534543Document559 pagesProg 4534543Vasile TroianNo ratings yet
- Saa6d170e-5 HPCR Egr Sen00190-04Document415 pagesSaa6d170e-5 HPCR Egr Sen00190-04Ahmad Mubarok100% (4)
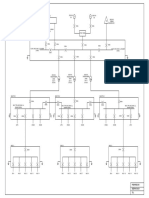
- Conceptual SLDDocument1 pageConceptual SLDakhilNo ratings yet
- Leptospirosis National Guidelines - Sri LankaDocument56 pagesLeptospirosis National Guidelines - Sri LankaBrainy-Paykiesaurus LuminirexNo ratings yet
- Nike Run Club 5K Training PlanDocument25 pagesNike Run Club 5K Training PlanSalvador0% (1)
- Energy Conversion and Management: Gvidonas Labeckas, Stasys Slavinskas, Irena KanapkieneDocument25 pagesEnergy Conversion and Management: Gvidonas Labeckas, Stasys Slavinskas, Irena KanapkieneVỵ ĐặngNo ratings yet
- To Study The Open Circuit/Core Losses of Single Phase TransformerDocument5 pagesTo Study The Open Circuit/Core Losses of Single Phase TransformerTanzeel UR RehmanNo ratings yet
- Specifications Models 37R116, 37R118 and 43R175: Carry Capacity: 18,200 To 47,200 Lbs (8250 To 21,400 KG.)Document7 pagesSpecifications Models 37R116, 37R118 and 43R175: Carry Capacity: 18,200 To 47,200 Lbs (8250 To 21,400 KG.)Marek WyszatyckiNo ratings yet
- Production Choke BasicsDocument39 pagesProduction Choke Basicsbtmohamed7084100% (1)
- Pump CommissioningDocument1 pagePump CommissioningMD SAMANNo ratings yet
- Dolphin Facts For Kids Ilovepdf CompressedDocument1 pageDolphin Facts For Kids Ilovepdf CompressedtechboostmrktgNo ratings yet
- Mater BiDocument10 pagesMater BihanjunyieeNo ratings yet
- Australian F1 SUBMACHINE GUNDocument4 pagesAustralian F1 SUBMACHINE GUNCaprikorn100% (4)
- Asme Section II A Sa-435 Sa-435mDocument4 pagesAsme Section II A Sa-435 Sa-435mAnonymous GhPzn1xNo ratings yet
- Proposal For Mining Skill TrainingDocument31 pagesProposal For Mining Skill Trainingdwarka prasad100% (1)
- Infographic Registration of Pharmaceutical Product For General Sale 637828524496205508Document1 pageInfographic Registration of Pharmaceutical Product For General Sale 637828524496205508Kdp03No ratings yet
- Biosystems Engineering BS CurriculumDocument2 pagesBiosystems Engineering BS CurriculumAsiiimweNo ratings yet