You might also like
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Lesson 3 Technical SketchingDocument39 pagesLesson 3 Technical SketchingMark Anthony LegaspiNo ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Characteristics of Water Quality of Municipal Wastewater Treatment Plants in China: Implications For Resources Utilization and ManagementDocument21 pagesCharacteristics of Water Quality of Municipal Wastewater Treatment Plants in China: Implications For Resources Utilization and ManagementDien NoelNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
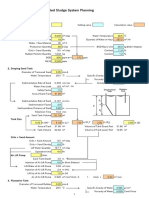
- Activated Sludge System PlanningDocument63 pagesActivated Sludge System PlanningSung Woong MoonNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Atmospheric Pollution Research: Faruk Dinçer, Fatih Kemal Dinçer, Deniz Sar I, Özcan Ceylan, Özgen ErcanDocument7 pagesAtmospheric Pollution Research: Faruk Dinçer, Fatih Kemal Dinçer, Deniz Sar I, Özcan Ceylan, Özgen ErcanAndres Felipe GuerraNo ratings yet
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
- Basic Construction - Lec1Document32 pagesBasic Construction - Lec1Dien NoelNo ratings yet
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- ppt02-1Document32 pagesppt02-1Dien NoelNo ratings yet
- Technical Drawing CH 05Document31 pagesTechnical Drawing CH 05Mohd Nizam ShakimonNo ratings yet
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Air CBE9312Document6 pagesAir CBE9312Dien NoelNo ratings yet
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- AirpolutionDocument4 pagesAirpolutionDien NoelNo ratings yet
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Modelling of The COD, TSS, Phosphate and Nitrate Distribution Due To The Sidoardjo Mud Flow Into Porong River EstuaryDocument8 pagesModelling of The COD, TSS, Phosphate and Nitrate Distribution Due To The Sidoardjo Mud Flow Into Porong River EstuarySuntoyo SajaNo ratings yet
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- ISSS AC Program (For Participants)Document3 pagesISSS AC Program (For Participants)Dien NoelNo ratings yet
- Program Book ICSBE 2022Document78 pagesProgram Book ICSBE 2022Dien NoelNo ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Amm 898 9Document8 pagesAmm 898 9Dien NoelNo ratings yet
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Wa TERDocument77 pagesWa TERDien NoelNo ratings yet
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Types of Research - Educational Research Basics by Del SiegleDocument3 pagesTypes of Research - Educational Research Basics by Del SiegleDien NoelNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Annex - 25 - Life Cycle Cost Comparison of Different STP ProcessesDocument10 pagesAnnex - 25 - Life Cycle Cost Comparison of Different STP ProcessesDien NoelNo ratings yet
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Presentationgo: Insert Your Picture Here (Click The Icon) and Send This Placeholder To BackDocument3 pagesPresentationgo: Insert Your Picture Here (Click The Icon) and Send This Placeholder To BackDien NoelNo ratings yet
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- Example AcademicDocument4 pagesExample AcademicSandy Indra PNo ratings yet
- EC ChallengesDocument18 pagesEC ChallengesMaliha CheemaNo ratings yet
- Modelling of The COD, TSS, Phosphate and Nitrate Distribution Due To The Sidoardjo Mud Flow Into Porong River EstuaryDocument8 pagesModelling of The COD, TSS, Phosphate and Nitrate Distribution Due To The Sidoardjo Mud Flow Into Porong River EstuarySuntoyo SajaNo ratings yet
- ADHD and Conduct Disorder: An MRI Study in A Community SampleDocument6 pagesADHD and Conduct Disorder: An MRI Study in A Community SampleDien NoelNo ratings yet
- Psychoradiologic Utility of MR Imaging For Diagnosis of Attention Deficit Hyperactivity DisorderDocument11 pagesPsychoradiologic Utility of MR Imaging For Diagnosis of Attention Deficit Hyperactivity DisorderDien NoelNo ratings yet
- Hessd 8 C5819 2012 SupplementDocument15 pagesHessd 8 C5819 2012 SupplementDien NoelNo ratings yet
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Using QUAL2Kw As A Decision Support Tool - Considerations For DataDocument110 pagesUsing QUAL2Kw As A Decision Support Tool - Considerations For DataAhmad Zaky NugrahaNo ratings yet
- New objective function for water balance reconciliationDocument6 pagesNew objective function for water balance reconciliationDien NoelNo ratings yet
- Ipi271951 PDFDocument8 pagesIpi271951 PDFjoana12No ratings yet
- Winrip Doc Larap Study Larap Bts. Kota Pariaman Manggopoh 20150918 00282Document66 pagesWinrip Doc Larap Study Larap Bts. Kota Pariaman Manggopoh 20150918 00282Dien NoelNo ratings yet
- Flood RescueDocument7 pagesFlood RescueDien NoelNo ratings yet
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Fenner2007 PDFDocument13 pagesFenner2007 PDFDien NoelNo ratings yet
- Chemosphere: D.J. Batstone, T. Hülsen, C.M. Mehta, J. KellerDocument10 pagesChemosphere: D.J. Batstone, T. Hülsen, C.M. Mehta, J. KellerDien NoelNo ratings yet
- PSE Inc. V CA G.R. No. 125469, Oct 27, 1997Document7 pagesPSE Inc. V CA G.R. No. 125469, Oct 27, 1997mae ann rodolfoNo ratings yet
- Assignment 3-WEF-Global Competitive IndexDocument3 pagesAssignment 3-WEF-Global Competitive IndexNauman MalikNo ratings yet
- HSG Anh 9 Thanh Thuy 2 (2018-2019) .Document8 pagesHSG Anh 9 Thanh Thuy 2 (2018-2019) .Huệ MẫnNo ratings yet
- ThumbDocument32 pagesThumbdhapraNo ratings yet
- Safe Handling of Solid Ammonium Nitrate: Recommendations For The Environmental Management of Commercial ExplosivesDocument48 pagesSafe Handling of Solid Ammonium Nitrate: Recommendations For The Environmental Management of Commercial ExplosivesCuesta AndresNo ratings yet
- Certificate of Compliance ATF F 5330 20Document2 pagesCertificate of Compliance ATF F 5330 20Jojo Aboyme CorcillesNo ratings yet
- 2009 WORD White Paper TemplateDocument4 pages2009 WORD White Paper Templateomegalpha777No ratings yet
- Unit-2 Fourier Series & Integral: 2130002 - Advanced Engineering MathematicsDocument143 pagesUnit-2 Fourier Series & Integral: 2130002 - Advanced Engineering MathematicsDarji DhrutiNo ratings yet
- Leaflet STP2025 LightDocument2 pagesLeaflet STP2025 LightNoel AjocNo ratings yet
- Chapter 018Document12 pagesChapter 018api-281340024No ratings yet
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Apola Ose-Otura (Popoola PDFDocument2 pagesApola Ose-Otura (Popoola PDFHowe JosephNo ratings yet
- I Could Easily FallDocument3 pagesI Could Easily FallBenji100% (1)
- Topic 8 - Managing Early Growth of The New VentureDocument11 pagesTopic 8 - Managing Early Growth of The New VentureMohamad Amirul Azry Chow100% (3)
- Restructuring ScenariosDocument57 pagesRestructuring ScenariosEmir KarabegovićNo ratings yet
- Love in Plato's SymposiumDocument31 pagesLove in Plato's Symposiumac12788100% (2)
- Application of Neutralization Titrations for Acid-Base AnalysisDocument21 pagesApplication of Neutralization Titrations for Acid-Base AnalysisAdrian NavarraNo ratings yet
- Plusnet Cancellation FormDocument2 pagesPlusnet Cancellation FormJoJo GunnellNo ratings yet
- Mce Igcse Chemistry PPT c08Document57 pagesMce Igcse Chemistry PPT c08Shabanito GamingNo ratings yet
- OutletsDocument226 pagesOutletsPraveen Kumar Saini100% (1)
- BLUEBOOK CITATION GUIDEDocument12 pagesBLUEBOOK CITATION GUIDEMichaela PortarcosNo ratings yet
- The Conflict With Slavery and Others, Complete, Volume VII, The Works of Whittier: The Conflict With Slavery, Politicsand Reform, The Inner Life and Criticism by Whittier, John Greenleaf, 1807-1892Document180 pagesThe Conflict With Slavery and Others, Complete, Volume VII, The Works of Whittier: The Conflict With Slavery, Politicsand Reform, The Inner Life and Criticism by Whittier, John Greenleaf, 1807-1892Gutenberg.org100% (1)
- Society and CultureDocument40 pagesSociety and CultureRichard AbellaNo ratings yet
- PAPC - Internal Notes PDFDocument4 pagesPAPC - Internal Notes PDFHrushi Km GowdaNo ratings yet
- KNJN Fpga Pluto-P BoardDocument15 pagesKNJN Fpga Pluto-P Boardgandalf1024No ratings yet
- Situation AnalysisDocument94 pagesSituation Analysisamirafateha100% (2)
- D2 Pre-Board Prof. Ed. - Do Not Teach Too Many Subjects.Document11 pagesD2 Pre-Board Prof. Ed. - Do Not Teach Too Many Subjects.Jorge Mrose26No ratings yet
- AMA Manual 10th Edition PDFDocument1,014 pagesAMA Manual 10th Edition PDFKannan Fangs S100% (2)
- IAS 8 Tutorial Question (SS)Document2 pagesIAS 8 Tutorial Question (SS)Given RefilweNo ratings yet
- Toyota TPMDocument23 pagesToyota TPMchteo1976No ratings yet
- Criminal Evidence Course OutlineDocument3 pagesCriminal Evidence Course OutlineChivas Gocela Dulguime100% (1)
- Stuff Matters: Exploring the Marvelous Materials That Shape Our Man-Made WorldFrom EverandStuff Matters: Exploring the Marvelous Materials That Shape Our Man-Made WorldRating: 4 out of 5 stars4/5 (289)
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeFrom EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (3)
- Science Goes Viral: Captivating Accounts of Science in Everyday LifeFrom EverandScience Goes Viral: Captivating Accounts of Science in Everyday LifeRating: 5 out of 5 stars5/5 (1)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsFrom EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsRating: 4 out of 5 stars4/5 (146)
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolFrom EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolNo ratings yet
- An Introduction to the Periodic Table of Elements : Chemistry Textbook Grade 8 | Children's Chemistry BooksFrom EverandAn Introduction to the Periodic Table of Elements : Chemistry Textbook Grade 8 | Children's Chemistry BooksRating: 5 out of 5 stars5/5 (1)
- Guidelines for Asset Integrity ManagementFrom EverandGuidelines for Asset Integrity ManagementRating: 5 out of 5 stars5/5 (1)