You might also like
- 5 Sindromes Mas ComunesDocument3 pages5 Sindromes Mas ComunesHugo MartinezNo ratings yet
- 10 Enfermedades Congénitas y Síndromes HabitualesDocument9 pages10 Enfermedades Congénitas y Síndromes HabitualesSindi AcevedoNo ratings yet
- Sindrome de Treacher CollinsDocument11 pagesSindrome de Treacher CollinsVania PortugalNo ratings yet
- CICLOPIADocument5 pagesCICLOPIAElda ZuñigaNo ratings yet
- Síndrome de Goldenhar.Document4 pagesSíndrome de Goldenhar.Natalia Vázquez EspírituNo ratings yet
- Malformaicones Congenitas de OidoDocument17 pagesMalformaicones Congenitas de OidoJorge Alejandro Duarte QuiñonezNo ratings yet
- Clase Anomalias Desarrollo Estructuras Bucales y DentariasDocument87 pagesClase Anomalias Desarrollo Estructuras Bucales y DentariasSantos Perez TolentinoNo ratings yet
- SEMIOLOGÍADocument13 pagesSEMIOLOGÍAnelsonNo ratings yet
- Disostosis MandibulofacialDocument6 pagesDisostosis MandibulofacialChristian BermeoNo ratings yet
- Enfermedades Genéticas Que Afectan La Cavidad Bucal PDFDocument9 pagesEnfermedades Genéticas Que Afectan La Cavidad Bucal PDFIdalid VillegasNo ratings yet
- SíndromesDocument7 pagesSíndromesHelena ACNo ratings yet
- Malformaciones CongénitasDocument33 pagesMalformaciones CongénitasJonathan ChiroqueNo ratings yet
- Síndrome de Treacher-CollinsDocument10 pagesSíndrome de Treacher-CollinsMarco Morante VegaNo ratings yet
- Displasia CleidocranealDocument10 pagesDisplasia CleidocranealSecia Lizbeth Clara PalomaresNo ratings yet
- Enfermedades Genéticas SindromesDocument22 pagesEnfermedades Genéticas SindromesJose Ccahuana EscalanteNo ratings yet
- Treacher Collins - CrouzonDocument6 pagesTreacher Collins - CrouzonNeiba RomeroNo ratings yet
- Album de Anomalias de La Tercera Semana de DesarrolloDocument11 pagesAlbum de Anomalias de La Tercera Semana de DesarrolloCamilo PureNo ratings yet
- 2.-Clasificacion de Las Malformaciones CraneofacialesDocument9 pages2.-Clasificacion de Las Malformaciones Craneofacialesluis Americo DuranNo ratings yet
- ApertDocument16 pagesApertmaricriscarNo ratings yet
- Discapacidad Auditiva 1Document4 pagesDiscapacidad Auditiva 1Yesenia Garcia (Miss Yessy)No ratings yet
- Malformaciones Craneofaciales Relacionadas Con La Cresta NeuralDocument7 pagesMalformaciones Craneofaciales Relacionadas Con La Cresta NeuralLaura CarreñoNo ratings yet
- Malformaciones CongenitasDocument8 pagesMalformaciones Congenitasdarwin041080No ratings yet
- Patologías Embriologicas Del Miembro SuperiorDocument10 pagesPatologías Embriologicas Del Miembro SuperiorJefferson MundacaNo ratings yet
- Alteraciones de La Motricidad Orofacial 1Document12 pagesAlteraciones de La Motricidad Orofacial 1Diliannys S.No ratings yet
- Monografía - Síndrome de Marfan DefinitivoDocument14 pagesMonografía - Síndrome de Marfan DefinitivoSadith ArpasiNo ratings yet
- Tercer PP Ptologias Ob AndresDocument25 pagesTercer PP Ptologias Ob Andresapi-290513027No ratings yet
- Anomalías EmbriologicasDocument2 pagesAnomalías EmbriologicasCarlos Daniel Martínez OrozcoNo ratings yet
- Síndrome de Dawn AtenciónDocument13 pagesSíndrome de Dawn AtenciónRAQUEL ALDAMA UAEHNo ratings yet
- Clasificacion de SindromesDocument41 pagesClasificacion de SindromesRodrigoAdrianCuevasCanoNo ratings yet
- A Anomalías Del Desarrollo de CaraDocument13 pagesA Anomalías Del Desarrollo de CaraDianitaVelezPincayNo ratings yet
- Enfermedades Genéticas Que Afectan La Cavidad BucalDocument16 pagesEnfermedades Genéticas Que Afectan La Cavidad BucalJessiEliNo ratings yet
- Enfermedades Genéticas Que Afectan La Cavidad BucalDocument12 pagesEnfermedades Genéticas Que Afectan La Cavidad BucalMauricio VallejosNo ratings yet
- Discapacidades CongénitasDocument2 pagesDiscapacidades CongénitasArnoldo Geovany GarciaNo ratings yet
- ACONDROPLASIADocument40 pagesACONDROPLASIAMiguel Angel NovoaNo ratings yet
- Documento Treacher CollinsDocument4 pagesDocumento Treacher CollinsNancyNo ratings yet
- HistologíaDocument24 pagesHistologíaDian JimezNo ratings yet
- CiclopiaDocument13 pagesCiclopiaThaynnara Araújo HespporteNo ratings yet
- Sindromes BucalesDocument12 pagesSindromes BucalesAleyda Bonfil León100% (1)
- Anomalías Estructurales de Los CromosomasDocument5 pagesAnomalías Estructurales de Los CromosomasgenesisNo ratings yet
- Osteogénesis ImperfectaDocument39 pagesOsteogénesis ImperfectaJonnathan Lizcano PinzonNo ratings yet
- Malformaciones Dentofaciales en SíndromesDocument6 pagesMalformaciones Dentofaciales en SíndromesManuel SerranoNo ratings yet
- Sindromes MayDocument108 pagesSindromes MayMaylor Fernández Mendez100% (2)
- Parálisis FacialDocument10 pagesParálisis FacialPaula FuentesNo ratings yet
- Cromosoma 17Document16 pagesCromosoma 17Isidora Montalva MoragaNo ratings yet
- Síndrome de Goldenhar y HunterDocument3 pagesSíndrome de Goldenhar y HunterYerling MendozaNo ratings yet
- REsumen EFAV...Document4 pagesREsumen EFAV...Sam Lathrop BrownNo ratings yet
- Anormalidades y Síndromes Diagonales de Ojo, Oido y CaraDocument15 pagesAnormalidades y Síndromes Diagonales de Ojo, Oido y Cara3RHM 38 Ortiz mora Luz celesteNo ratings yet
- Diagnosticos Diferenciales de AnodonciaDocument8 pagesDiagnosticos Diferenciales de Anodonciashantal1zazuetaNo ratings yet
- Trabajo de Embriologia 1Document20 pagesTrabajo de Embriologia 1Elbio VasquezNo ratings yet
- Disostosis-Cleidocraneal FinaaaaaaaalDocument6 pagesDisostosis-Cleidocraneal FinaaaaaaaalKathe MorenoNo ratings yet
- Paladar Hendido Parcial y CompletoDocument13 pagesPaladar Hendido Parcial y CompletoMartin PerezNo ratings yet
- ProgeriaDocument6 pagesProgeriaJoseArteagaNo ratings yet
- Síndrome de Goldenhar.Document3 pagesSíndrome de Goldenhar.Yerling MendozaNo ratings yet
- MalformacionesDocument35 pagesMalformacionesmelani hernandezNo ratings yet
- 2022 Anomalias CraneofacialesDocument50 pages2022 Anomalias CraneofacialesAngie Acosta RodriguezNo ratings yet
- Síndrome de Ehlers DanlosDocument20 pagesSíndrome de Ehlers DanlosLeo DoctorhideNo ratings yet
- Síndrome de Treacher CollinsDocument2 pagesSíndrome de Treacher Collinsnicaragua pediatrica100% (2)
- Hipoacusias Perceptivas Postnatales Hereditarias Que Constituyen Síndromes PDFDocument5 pagesHipoacusias Perceptivas Postnatales Hereditarias Que Constituyen Síndromes PDFSONIA BEATRIZ BURGOA SILVANo ratings yet
- ExposiciònDocument21 pagesExposiciònprotesis fija 1No ratings yet
- Historia Clínica PediDocument16 pagesHistoria Clínica PediLiz Sofia UsamiNo ratings yet
- ArticulacionesDocument17 pagesArticulacionesLiz Sofia UsamiNo ratings yet
- HISTORIA CLINICA VaricelaDocument42 pagesHISTORIA CLINICA VaricelaLiz Sofia UsamiNo ratings yet
- PsoriasisDocument26 pagesPsoriasisLiz Sofia UsamiNo ratings yet
- Poe, Edgar Allan - El Corazon Delator (1843)Document0 pagesPoe, Edgar Allan - El Corazon Delator (1843)Martha RLNo ratings yet
- Adenocarcinoma Pulmonar Caso ClinicoDocument4 pagesAdenocarcinoma Pulmonar Caso ClinicoLiz Sofia UsamiNo ratings yet
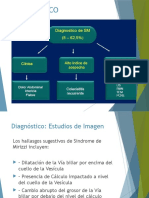
- Sindrome de Mirizzi Diagnostico y TratamientoDocument11 pagesSindrome de Mirizzi Diagnostico y TratamientoSofia DuranNo ratings yet
- CRIPTORQUIDIADocument6 pagesCRIPTORQUIDIALiz Sofia UsamiNo ratings yet
- Hipertension IntracraneanaDocument19 pagesHipertension IntracraneanaLiz Sofia UsamiNo ratings yet
- Rinosinusitis Cronica y Sus ComplicacionesDocument12 pagesRinosinusitis Cronica y Sus ComplicacionesLiz Sofia UsamiNo ratings yet
- PsoriasisDocument26 pagesPsoriasisLiz Sofia UsamiNo ratings yet
- Ataque Isquemico TransitorioDocument16 pagesAtaque Isquemico TransitorioLiz Sofia Usami100% (2)
- Seminario Osteomielitis CronicaDocument31 pagesSeminario Osteomielitis CronicaLiz Sofia UsamiNo ratings yet
- MarasmoDocument3 pagesMarasmoLiz Sofia UsamiNo ratings yet
- Formas Farmacéuticas y Vías de AdministraciónDocument65 pagesFormas Farmacéuticas y Vías de AdministraciónkellydiazromeroNo ratings yet
- Guía Elaboración Informe Final de La Investigación 2013Document7 pagesGuía Elaboración Informe Final de La Investigación 2013Liz Sofia UsamiNo ratings yet
- CASO CLINICO (Quiste Hidatidico)Document6 pagesCASO CLINICO (Quiste Hidatidico)Liz Sofia UsamiNo ratings yet
- Caso ClinicoDocument5 pagesCaso ClinicoLiz Sofia UsamiNo ratings yet
- LeishmaniasisDocument1 pageLeishmaniasisLiz Sofia UsamiNo ratings yet
- Fisiologia Aparato DigestivoDocument1 pageFisiologia Aparato DigestivoLiz Sofia UsamiNo ratings yet
- Alegra Tu VidaDocument2 pagesAlegra Tu Vidaisrael24640% (5)
- Maltrato AnimalDocument24 pagesMaltrato Animalmichecita78% (9)
- Nom 087 Ecol Ssa1 2002Document19 pagesNom 087 Ecol Ssa1 2002AgustínCamachoNo ratings yet
- El AguaDocument3 pagesEl AguaFernando PantaNo ratings yet
- Inen 1217Document8 pagesInen 1217Inesita CordovaNo ratings yet
- Guia Campo Sobre Tipos de Insectos y Acaros Que Infestan El Pescado CuradoDocument28 pagesGuia Campo Sobre Tipos de Insectos y Acaros Que Infestan El Pescado CuradoXochy EnsenadaNo ratings yet
- Seminario Grupo2Document5 pagesSeminario Grupo2Lizeth Benito RamosNo ratings yet
- C-1º ACT 4 CYT UNID 3 - SEM 3 (Autoguardado)Document5 pagesC-1º ACT 4 CYT UNID 3 - SEM 3 (Autoguardado)Teobaldo QuevedoNo ratings yet
- Las ArañasDocument7 pagesLas Arañasscribo88No ratings yet
- Tratamiento de La Oclusion Arterial AgudaDocument5 pagesTratamiento de La Oclusion Arterial AgudaAna BepNo ratings yet
- Sistema Excretor en Animales y HumanosDocument9 pagesSistema Excretor en Animales y HumanosJulia Peralta MeloNo ratings yet
- 1 Salado de PescadoDocument3 pages1 Salado de PescadoJose Martin Nuñez PereyraNo ratings yet
- S02.s1 - Material. Medio Ambiente y EcosistemaDocument30 pagesS02.s1 - Material. Medio Ambiente y EcosistemaKatherine MfjNo ratings yet
- Métodos y Técnicas en Etología AnimalDocument26 pagesMétodos y Técnicas en Etología AnimalG ColleenNo ratings yet
- Resumen Adulto MayorDocument25 pagesResumen Adulto MayorJuan SepulvedaNo ratings yet
- Neumonía y EAP: Diagnostico RadiológicoDocument28 pagesNeumonía y EAP: Diagnostico RadiológicoJorge Gonzales Paredes50% (2)
- Ecbi 2° Ciclos de VidaDocument8 pagesEcbi 2° Ciclos de VidaPatricio Solis HuiriqueoNo ratings yet
- Sistema Nervioso HumanoDocument3 pagesSistema Nervioso HumanoNahir ZentenoNo ratings yet
- Anticuerpos Antineuronales ParaneoplásicosDocument2 pagesAnticuerpos Antineuronales ParaneoplásicosfeliprolNo ratings yet
- Instructivo EscoltasDocument22 pagesInstructivo EscoltasLaura Rb100% (1)
- Biomas, Bosques y FaunaDocument39 pagesBiomas, Bosques y FaunaLisbeth Arias SuarezNo ratings yet
- AMEBIASISDocument13 pagesAMEBIASISHazzel RiveraNo ratings yet
- Resistencia Del Organismo A La Infección, Inunidad y AlergiaDocument4 pagesResistencia Del Organismo A La Infección, Inunidad y Alergiaemtaguirre100% (1)
- Sindrome de ReiterDocument2 pagesSindrome de ReiterIrvin SGNo ratings yet
- Tecnicas de Examen GinecologicoDocument123 pagesTecnicas de Examen GinecologicoUlises MonteonNo ratings yet
- Estructuras Sensoriales de Las ArteriasDocument1 pageEstructuras Sensoriales de Las ArteriasMaca SandovalNo ratings yet
- MEDICIONES RADIOGRAFICAS. Equipo Arturoleyva, Ivanparada, DiegolimonDocument48 pagesMEDICIONES RADIOGRAFICAS. Equipo Arturoleyva, Ivanparada, DiegolimonArturo Leyva RiveraNo ratings yet
- ANPHIBIADocument7 pagesANPHIBIADiego Raul Ayquipa PonceNo ratings yet
- GUIA 2 - La Reproducción en AnimalesDocument8 pagesGUIA 2 - La Reproducción en AnimalesPapeleria telaraña escolarNo ratings yet
- Adaptacion Fisiologica Del Recien Nacido Al NacimientoDocument11 pagesAdaptacion Fisiologica Del Recien Nacido Al NacimientoCarmen Alejos RospigliosiNo ratings yet