You might also like
- Asset Intelligence Report: A Primer On Corrosion Under Insulation (CUI)Document5 pagesAsset Intelligence Report: A Primer On Corrosion Under Insulation (CUI)dzamir203No ratings yet
- Form of Corrosion Illustration Form of Corrosion IllustrationDocument2 pagesForm of Corrosion Illustration Form of Corrosion IllustrationkhalesnabilNo ratings yet
- Corrosion: Corrosion ControlFrom EverandCorrosion: Corrosion ControlL L ShreirRating: 5 out of 5 stars5/5 (1)
- Article CO2CorrosionCHEM409 - Background of CO2 CorrosionDocument4 pagesArticle CO2CorrosionCHEM409 - Background of CO2 Corrosionmohamed samyNo ratings yet
- API RP 571 Damage Mechanisms SpreadsheetDocument12 pagesAPI RP 571 Damage Mechanisms SpreadsheetSoftware ManagerNo ratings yet
- Biological Treatment of Microbial Corrosion: Opportunities and ChallengesFrom EverandBiological Treatment of Microbial Corrosion: Opportunities and ChallengesNo ratings yet
- Incoloy Alloy 25-6MODocument13 pagesIncoloy Alloy 25-6MOsiswoutNo ratings yet
- Corrosion and Materials in Hydrocarbon Production: A Compendium of Operational and Engineering AspectsFrom EverandCorrosion and Materials in Hydrocarbon Production: A Compendium of Operational and Engineering AspectsNo ratings yet
- CLSCC LiteratureDocument62 pagesCLSCC LiteratureNakarin PotidokmaiNo ratings yet
- Comparision Table For AluminumDocument2 pagesComparision Table For AluminumJigar M. UpadhyayNo ratings yet
- MIC in Heat Exchanger TubingDocument6 pagesMIC in Heat Exchanger TubingdutuconstantinNo ratings yet
- 0700testing MultipleDocument5 pages0700testing MultipleIvan GutierrezNo ratings yet
- Corrosion Quiz AnswersDocument5 pagesCorrosion Quiz AnswersErik Alfiandy100% (1)
- Centralloy G 4852 Micro R: Material Data SheetDocument8 pagesCentralloy G 4852 Micro R: Material Data SheetNest NectureNo ratings yet
- Grade 2205 DuplexDocument9 pagesGrade 2205 Duplexkresimir.mikoc9765No ratings yet
- Corrosion Problems in The Oil IndustryDocument8 pagesCorrosion Problems in The Oil IndustryUNIISCRIBDNo ratings yet
- CuiDocument6 pagesCuiأحمد صبحىNo ratings yet
- Mechanical FatigueDocument6 pagesMechanical FatigueRamyMoustafaNo ratings yet
- Bacterial Corrosion BDocument6 pagesBacterial Corrosion BNatrajiNo ratings yet
- Pitting Corrosion: A Localized Form of Corrosive AttackDocument7 pagesPitting Corrosion: A Localized Form of Corrosive AttackMuhammad Shena Gumilang100% (1)
- EFC No.62-2012 Testing Tribocorrosion of Passivating Materials Supporting ResearDocument215 pagesEFC No.62-2012 Testing Tribocorrosion of Passivating Materials Supporting ResearWHWENNo ratings yet
- Api 571 PDFDocument3 pagesApi 571 PDFumnartkhNo ratings yet
- Corrosion Resistance of High Nitrogen Steels PDFDocument27 pagesCorrosion Resistance of High Nitrogen Steels PDFAnil Kumar TNo ratings yet
- Corrosion and Runoff Behavior of Carbon Steel in Simulated Acid RainDocument3 pagesCorrosion and Runoff Behavior of Carbon Steel in Simulated Acid RainIvan GutierrezNo ratings yet
- Corrosion Test PiecesDocument23 pagesCorrosion Test PiecesGijoNo ratings yet
- Smarter Materials Selection For Corrosion Control PDFDocument12 pagesSmarter Materials Selection For Corrosion Control PDFAsyraf Nordin100% (1)
- AL 6XN SourceBookDocument56 pagesAL 6XN SourceBookdrbeyerNo ratings yet
- Flow in Flow-Accelerated Corrosion For Nuclear Power PlantDocument16 pagesFlow in Flow-Accelerated Corrosion For Nuclear Power PlantSalih Gürkan üyümezNo ratings yet
- Austenitic High Temperature 153MA 253MA Stainless BrochureDocument16 pagesAustenitic High Temperature 153MA 253MA Stainless BrochurezosternatNo ratings yet
- Corrosion Under Insulation - Out of Sight Out of MindDocument4 pagesCorrosion Under Insulation - Out of Sight Out of Mindengr_ahmednassarNo ratings yet
- Metallurgical Failure AnalysisDocument4 pagesMetallurgical Failure AnalysisgirishnitwNo ratings yet
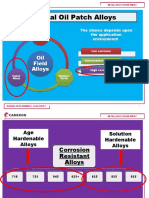
- Oil Field Alloy Selection GuideDocument11 pagesOil Field Alloy Selection GuideAnonymous 7yN43wjlNo ratings yet
- Corrosion Management Issue151Document32 pagesCorrosion Management Issue151Issam Mokrani100% (1)
- CSWIP NotesDocument6 pagesCSWIP Notestulasirao.nammiNo ratings yet
- T 18Document10 pagesT 18khuramluck100% (2)
- PTA SCC: Polythionic acid stress corrosion crackingDocument9 pagesPTA SCC: Polythionic acid stress corrosion crackingMedina EldesNo ratings yet
- Corrosion Behavior of Steels in Gulf SeawaterDocument15 pagesCorrosion Behavior of Steels in Gulf SeawaterDoctorAtomicNo ratings yet
- The Forms of Corrosion-Part2Document71 pagesThe Forms of Corrosion-Part2quiron2010100% (1)
- Cathodic Disbondment PDFDocument25 pagesCathodic Disbondment PDFsanjayaNo ratings yet
- ROOKWOOL (Non Contact Insulation)Document28 pagesROOKWOOL (Non Contact Insulation)devangmajithiaNo ratings yet
- Pipeline Stress Corrosion CrackingDocument7 pagesPipeline Stress Corrosion CrackingWendy AguilarNo ratings yet
- Hydrogen Induced Cracking PDFDocument254 pagesHydrogen Induced Cracking PDFAnas PratamaNo ratings yet
- Caustic Stress Corrosion Cracking of A Graphite Cast Iron ComponentDocument8 pagesCaustic Stress Corrosion Cracking of A Graphite Cast Iron Componentriza9847No ratings yet
- Microstructural Changes in Austenitic Stainless Steels During Long Term Aging. Minami1986 PDFDocument12 pagesMicrostructural Changes in Austenitic Stainless Steels During Long Term Aging. Minami1986 PDFAndrea CalderaNo ratings yet
- An Electrochemical Study of Cathodic Protection of Steel Used For Marine StructuresDocument6 pagesAn Electrochemical Study of Cathodic Protection of Steel Used For Marine StructureshamidNo ratings yet
- Environment Assisted Cracking ME 472: Corrosion EngineeringDocument45 pagesEnvironment Assisted Cracking ME 472: Corrosion EngineeringEmmanuelNo ratings yet
- Corrosion PDFDocument46 pagesCorrosion PDFNixon RamsaranNo ratings yet
- Sulfuric Acid and Hydrochloric Acid Dew-Point Corrosion-Resistant SteelDocument0 pagesSulfuric Acid and Hydrochloric Acid Dew-Point Corrosion-Resistant SteelMatt AgonyaNo ratings yet
- CORROSION QUIZ - InG - Painting and CoatingDocument3 pagesCORROSION QUIZ - InG - Painting and CoatingErik Alfiandy100% (1)
- CUI: An In-Depth Analysis: Hira S. AhluwaliaDocument6 pagesCUI: An In-Depth Analysis: Hira S. AhluwaliaJithuJohnNo ratings yet
- Hydrogen Cracking in Duplex Stainless Steel Weld MetalDocument10 pagesHydrogen Cracking in Duplex Stainless Steel Weld MetalOlgalycosNo ratings yet
- A Study of Corrosion Rate of Stainless Steels AISI 316 and 306 Against HCL H2SO4 and Dead Sea WaterDocument45 pagesA Study of Corrosion Rate of Stainless Steels AISI 316 and 306 Against HCL H2SO4 and Dead Sea WatermohdghNo ratings yet
- CH 06 - Corrosion & ErosionDocument22 pagesCH 06 - Corrosion & ErosionvegaronNo ratings yet
- Thermal Spray Coating Procection Steel PilingDocument169 pagesThermal Spray Coating Procection Steel Pilingdevancdm100% (1)
- Corrosion PDFDocument22 pagesCorrosion PDFVishal ThakareNo ratings yet
- 1 - Design and Analysis of Impeller Blade For Axial Flow PumpsDocument31 pages1 - Design and Analysis of Impeller Blade For Axial Flow PumpsmghgolNo ratings yet
- CAD Procedure for Blade Design of Centrifugal Pump ImpellerDocument8 pagesCAD Procedure for Blade Design of Centrifugal Pump ImpellermghgolNo ratings yet
- Eiraj Miraza's Divan of Poetry from Kalkade.comDocument0 pagesEiraj Miraza's Divan of Poetry from Kalkade.commeysamshabnavaaNo ratings yet
- Jsir 72 (6) 373-378Document6 pagesJsir 72 (6) 373-378mghgolNo ratings yet
- Surface Coating Operations MetalsDocument66 pagesSurface Coating Operations MetalsRaj Patel100% (1)
- JP C Purdue Pump PaperDocument15 pagesJP C Purdue Pump PapermghgolNo ratings yet
- Suri Rotor BalancingDocument55 pagesSuri Rotor BalancingDhaval BhayaniNo ratings yet
- Pump RotordynamicsDocument26 pagesPump RotordynamicsHalil İbrahim Küplü100% (1)
- Aiaa 2011 5932Document7 pagesAiaa 2011 5932mghgolNo ratings yet
- 1001 Shab 1Document268 pages1001 Shab 1ozmeraNo ratings yet
- p0155 PDFDocument9 pagesp0155 PDFAndrey ZagorulkoNo ratings yet
- Liquid-Oxygen Turbopumps: NASA TechnicalDocument16 pagesLiquid-Oxygen Turbopumps: NASA TechnicalmghgolNo ratings yet
- Aiaa 2011 1938Document13 pagesAiaa 2011 1938mghgolNo ratings yet
- Acoustics Based Condition MonitoringDocument7 pagesAcoustics Based Condition MonitoringDeniz YazgaçNo ratings yet
- 高瞻計畫 - 振動學課程 Lecture 1: Single Degree of Freedom Systems (III)Document29 pages高瞻計畫 - 振動學課程 Lecture 1: Single Degree of Freedom Systems (III)mghgolNo ratings yet
- Rtiwari RD Book 09Document21 pagesRtiwari RD Book 09mghgolNo ratings yet
- Rtiwari RD Book 06bDocument15 pagesRtiwari RD Book 06bmghgolNo ratings yet
- 126 ElamyDocument15 pages126 ElamymghgolNo ratings yet
- Rtiwari RD Book 10Document33 pagesRtiwari RD Book 10mghgolNo ratings yet
- Rtiwari RD Book 12Document21 pagesRtiwari RD Book 12mghgolNo ratings yet
- Rtiwari RD Book 11cDocument8 pagesRtiwari RD Book 11cmghgolNo ratings yet
- Rtiwari RD Book 06cDocument18 pagesRtiwari RD Book 06cmghgolNo ratings yet
- Rtiwari RD Book 05bDocument12 pagesRtiwari RD Book 05bmghgolNo ratings yet
- Rtiwari RD Book 11aDocument16 pagesRtiwari RD Book 11amghgolNo ratings yet
- Rtiwari RD Book 05aDocument22 pagesRtiwari RD Book 05amghgolNo ratings yet
- Rtiwari RD Book 08Document17 pagesRtiwari RD Book 08mghgolNo ratings yet
- Rtiwari RD Book 09Document21 pagesRtiwari RD Book 09mghgolNo ratings yet
- Rtiwari RD Book 03cDocument29 pagesRtiwari RD Book 03cmghgolNo ratings yet
- Rtiwari RD Book 03bDocument17 pagesRtiwari RD Book 03bmghgolNo ratings yet
- Adicat-01Document4 pagesAdicat-01hse indacoNo ratings yet
- Final Summative Exam #2 Grade 7Document4 pagesFinal Summative Exam #2 Grade 7Mae CudalNo ratings yet
- Alka-Seltzer PH DemoDocument2 pagesAlka-Seltzer PH Demoandycapo123No ratings yet
- SpectraLOCK PRO Grout PDFDocument3 pagesSpectraLOCK PRO Grout PDFFachuDARNo ratings yet
- Analysis of Fruit and Vegetable JuicesDocument3 pagesAnalysis of Fruit and Vegetable JuicesAditya Rajendran50% (2)
- CHM 3301 Lab Report Structure SolidsDocument7 pagesCHM 3301 Lab Report Structure SolidsAida NordinNo ratings yet
- Design of Gating and Riser System For Grate Bar CastingDocument6 pagesDesign of Gating and Riser System For Grate Bar CastingvaseaNo ratings yet
- B12 Series 2-wire Gas TransmittersDocument1 pageB12 Series 2-wire Gas TransmittersKareem RMGNo ratings yet
- Abosrption and Flammability Test On Banana LeafDocument6 pagesAbosrption and Flammability Test On Banana LeafsudhirNo ratings yet
- IB Chemistry: Unit 4 Stoichiometry QuestionsDocument37 pagesIB Chemistry: Unit 4 Stoichiometry QuestionsmjohnmccNo ratings yet
- Protic Ionic Liquids Properties and ApplicationsDocument32 pagesProtic Ionic Liquids Properties and Applicationsion.liqNo ratings yet
- Dafus PenilitianDocument5 pagesDafus PenilitianDim YotaNo ratings yet
- Din 50979Document14 pagesDin 50979Bangali Singh100% (3)
- Gasguard t2 Garments DatasheetDocument2 pagesGasguard t2 Garments Datasheetyongqian guoNo ratings yet
- Experimental Study On A Domestic Refrigerator Using LPG As A RefrigarantDocument8 pagesExperimental Study On A Domestic Refrigerator Using LPG As A RefrigarantAman KumarNo ratings yet
- Blasting and PaintingDocument64 pagesBlasting and PaintingSyahril Aizal Ahmad75% (4)
- Astm D 6386-99Document4 pagesAstm D 6386-99Mritunjay100% (2)
- Autogenous Healing of Cement Mortar and Concrete: Technical SeriesDocument4 pagesAutogenous Healing of Cement Mortar and Concrete: Technical Series张迪No ratings yet
- Caustic EmbrittlementDocument2 pagesCaustic Embrittlementrao9990No ratings yet
- PDS STRATA EPOXY HT 35590 en-GB PDFDocument3 pagesPDS STRATA EPOXY HT 35590 en-GB PDFMohamed Nouzer0% (1)
- Goc PDFDocument56 pagesGoc PDFManik SinghalNo ratings yet
- Water From The Surrounding Solution Due To Osmosis. The Process of Osmosis CausesDocument1 pageWater From The Surrounding Solution Due To Osmosis. The Process of Osmosis CausesMatthew BrownNo ratings yet
- Ingot ManufacturingDocument7 pagesIngot ManufacturingRamesh KuppiliNo ratings yet
- Lipid Extraction Basics of MTBE MethodDocument3 pagesLipid Extraction Basics of MTBE MethodCarla Elize DerainNo ratings yet
- Chapter 2 - Fundemental of MeterialsDocument84 pagesChapter 2 - Fundemental of Meterialsquan quanNo ratings yet
- Baroid Fluids HandbookDocument8 pagesBaroid Fluids HandbookTamer Hesham AhmedNo ratings yet
- Inorganic Chem 11Document68 pagesInorganic Chem 11hisrom286No ratings yet
- Unilever Australia MSDSDocument5 pagesUnilever Australia MSDSarditNo ratings yet
- Inconel 600 Alloy 600 Uns n06600 Din 2.4816Document3 pagesInconel 600 Alloy 600 Uns n06600 Din 2.4816SamkitNo ratings yet
- Liquid Densities kg/cu.mDocument4 pagesLiquid Densities kg/cu.mhenrytolentinoNo ratings yet
- Piping and Pipeline Calculations Manual: Construction, Design Fabrication and ExaminationFrom EverandPiping and Pipeline Calculations Manual: Construction, Design Fabrication and ExaminationRating: 4 out of 5 stars4/5 (18)
- Nuclear Energy in the 21st Century: World Nuclear University PressFrom EverandNuclear Energy in the 21st Century: World Nuclear University PressRating: 4.5 out of 5 stars4.5/5 (3)
- Functional Safety from Scratch: A Practical Guide to Process Industry ApplicationsFrom EverandFunctional Safety from Scratch: A Practical Guide to Process Industry ApplicationsNo ratings yet
- An Introduction to the Periodic Table of Elements : Chemistry Textbook Grade 8 | Children's Chemistry BooksFrom EverandAn Introduction to the Periodic Table of Elements : Chemistry Textbook Grade 8 | Children's Chemistry BooksRating: 5 out of 5 stars5/5 (1)
- Chemical Process Safety: Learning from Case HistoriesFrom EverandChemical Process Safety: Learning from Case HistoriesRating: 4 out of 5 stars4/5 (14)
- Guidelines for the Management of Change for Process SafetyFrom EverandGuidelines for the Management of Change for Process SafetyNo ratings yet
- Operational Excellence: Journey to Creating Sustainable ValueFrom EverandOperational Excellence: Journey to Creating Sustainable ValueNo ratings yet
- Process Engineering for a Small Planet: How to Reuse, Re-Purpose, and Retrofit Existing Process EquipmentFrom EverandProcess Engineering for a Small Planet: How to Reuse, Re-Purpose, and Retrofit Existing Process EquipmentNo ratings yet
- Guidelines for Siting and Layout of FacilitiesFrom EverandGuidelines for Siting and Layout of FacilitiesNo ratings yet
- Well Control for Completions and InterventionsFrom EverandWell Control for Completions and InterventionsRating: 4 out of 5 stars4/5 (10)
- Guidelines for Developing Quantitative Safety Risk CriteriaFrom EverandGuidelines for Developing Quantitative Safety Risk CriteriaNo ratings yet
- Guidelines for Enabling Conditions and Conditional Modifiers in Layer of Protection AnalysisFrom EverandGuidelines for Enabling Conditions and Conditional Modifiers in Layer of Protection AnalysisNo ratings yet
- Guidelines for Engineering Design for Process SafetyFrom EverandGuidelines for Engineering Design for Process SafetyNo ratings yet
- Lees' Process Safety Essentials: Hazard Identification, Assessment and ControlFrom EverandLees' Process Safety Essentials: Hazard Identification, Assessment and ControlRating: 4 out of 5 stars4/5 (4)
- Perfume Engineering: Design, Performance and ClassificationFrom EverandPerfume Engineering: Design, Performance and ClassificationRating: 4 out of 5 stars4/5 (5)
- Practical Process Control for Engineers and TechniciansFrom EverandPractical Process Control for Engineers and TechniciansRating: 5 out of 5 stars5/5 (3)
- Handbook of Cosmetic Science: An Introduction to Principles and ApplicationsFrom EverandHandbook of Cosmetic Science: An Introduction to Principles and ApplicationsH. W. HibbottRating: 4 out of 5 stars4/5 (6)
- Process Plant Equipment: Operation, Control, and ReliabilityFrom EverandProcess Plant Equipment: Operation, Control, and ReliabilityRating: 5 out of 5 stars5/5 (1)
- Bow Ties in Risk Management: A Concept Book for Process SafetyFrom EverandBow Ties in Risk Management: A Concept Book for Process SafetyNo ratings yet
- Process Steam Systems: A Practical Guide for Operators, Maintainers, and DesignersFrom EverandProcess Steam Systems: A Practical Guide for Operators, Maintainers, and DesignersNo ratings yet
- Fragrance Chemistry: The Science of the Sense of SmellFrom EverandFragrance Chemistry: The Science of the Sense of SmellRating: 3 out of 5 stars3/5 (2)