You might also like
- X-Ray Crystallography and Its Applications in Dairy Science: A ReviewDocument13 pagesX-Ray Crystallography and Its Applications in Dairy Science: A Reviewkamal gandhiNo ratings yet
- Comparison of Crystallography, NMR, cryoEMDocument9 pagesComparison of Crystallography, NMR, cryoEMHằngHamHốNo ratings yet
- X Ray Crystallography and Its ApplicatioDocument13 pagesX Ray Crystallography and Its ApplicatioManoj PrakashNo ratings yet
- Catalyst Characterization TechniquesDocument8 pagesCatalyst Characterization TechniquesDaniel DadzieNo ratings yet
- RmnproteinasDocument13 pagesRmnproteinasfc99No ratings yet
- Techniques For Characterization of Nano MaterialsDocument35 pagesTechniques For Characterization of Nano Materialsnur_rizqi_1No ratings yet
- 04 X-Ray DiffractionDocument9 pages04 X-Ray DiffractionSampath KumarNo ratings yet
- Characterization of Nanomaterials - Ch3-Student 2020 PDFDocument12 pagesCharacterization of Nanomaterials - Ch3-Student 2020 PDFWasan ShakirNo ratings yet
- HighOrderAberrations02-Zivanov 2020Document15 pagesHighOrderAberrations02-Zivanov 2020Tongyi DouNo ratings yet
- 5 STR DeterminationTech 5oct2020Document22 pages5 STR DeterminationTech 5oct2020things strangerNo ratings yet
- Lab Mst613 (Part B)Document10 pagesLab Mst613 (Part B)hyebibieNo ratings yet
- Utilizing Web-Based Search Engines for Analyzing Biological MacromoleculesFrom EverandUtilizing Web-Based Search Engines for Analyzing Biological MacromoleculesNo ratings yet
- MicroscopeDocument3 pagesMicroscoperuskaNo ratings yet
- Reading Paper 2Document11 pagesReading Paper 2Yueyue ZhaoNo ratings yet
- CoppensP Experimental JACS1999Document9 pagesCoppensP Experimental JACS1999bubczenkoNo ratings yet
- Journal of Molecular StructureDocument11 pagesJournal of Molecular StructureRadhikaNo ratings yet
- Ncomms 14999Document8 pagesNcomms 14999jawad aliNo ratings yet
- Direct Assignment of Molecular Vibrations Via Normal Mode Analysis of The Neutron Dynamic Pair Distribution Function TechniqueDocument24 pagesDirect Assignment of Molecular Vibrations Via Normal Mode Analysis of The Neutron Dynamic Pair Distribution Function TechniqueRabbia AminNo ratings yet
- Yuri A. Gruzdkov and Yogendra M. Gupta - Vibrational Properties and Structure of Pentaerythritol TetranitrateDocument6 pagesYuri A. Gruzdkov and Yogendra M. Gupta - Vibrational Properties and Structure of Pentaerythritol TetranitratePomaxxNo ratings yet
- Spectroscopy TechniquesDocument3 pagesSpectroscopy TechniquesSantosh prajapatiNo ratings yet
- Small-Angle Scattering: A View On The Properties, Structures and Structural Changes of Biological Macromolecules in SolutionDocument81 pagesSmall-Angle Scattering: A View On The Properties, Structures and Structural Changes of Biological Macromolecules in SolutiondibudkNo ratings yet
- J. N. Reddy - 2022 - Tridynamic Model of The Beam With Transverse Shear DeformationDocument20 pagesJ. N. Reddy - 2022 - Tridynamic Model of The Beam With Transverse Shear DeformationSevim GüçlüNo ratings yet
- Byler 1988Document16 pagesByler 1988Amuthachelvi DanielNo ratings yet
- 2021 MM 05Document8 pages2021 MM 05Ayyan AnwarNo ratings yet
- Said ReviewDocument13 pagesSaid Reviewlab sopsNo ratings yet
- SLSPDFDocument4 pagesSLSPDFVivek BelaNo ratings yet
- Molecules: An Overview of Molecular Modeling For Drug Discovery With Specific Illustrative Examples of ApplicationsDocument32 pagesMolecules: An Overview of Molecular Modeling For Drug Discovery With Specific Illustrative Examples of ApplicationsJose David Perez NavarroNo ratings yet
- Voltametría de Micropartículas en La Diferenciación de Óxidos y Oxihidróxidos de HierroDocument2 pagesVoltametría de Micropartículas en La Diferenciación de Óxidos y Oxihidróxidos de HierroMileisi Fernández CarrascoNo ratings yet
- Preparation of Dichlorobis - (Ethylenediamine) Cobalt (Iii) Chloride and Characterization With Single Crystal X-Ray DiffractionDocument7 pagesPreparation of Dichlorobis - (Ethylenediamine) Cobalt (Iii) Chloride and Characterization With Single Crystal X-Ray DiffractionJ Mora GañanNo ratings yet
- jp993593c PDFDocument23 pagesjp993593c PDFmbolantenainaNo ratings yet
- TOPIC 15E Spectroscopy and ChromatographyDocument30 pagesTOPIC 15E Spectroscopy and ChromatographyAyshath MaaishaNo ratings yet
- McCulloch Et Al. - 2000 - Ab Initio Simulations of The Structure of AmorphouDocument7 pagesMcCulloch Et Al. - 2000 - Ab Initio Simulations of The Structure of AmorphouAlireza BagherpourNo ratings yet
- Electronic G Tensors in U Complexes-A Computational Study: DOI: 10.1002/chem.201701058Document11 pagesElectronic G Tensors in U Complexes-A Computational Study: DOI: 10.1002/chem.201701058Austin LloydNo ratings yet
- Nuclear Instruments and Methods in Physics Research A: Irène BuvatDocument7 pagesNuclear Instruments and Methods in Physics Research A: Irène BuvatKhairulNo ratings yet
- Powder DiffractionDocument22 pagesPowder DiffractionTamara Silvana CárcamoNo ratings yet
- Characterization Techniques 6.1 Scanning Electron Microscope (Sem)Document7 pagesCharacterization Techniques 6.1 Scanning Electron Microscope (Sem)RanNo ratings yet
- PHD Thesis Surface Plasmon ResonanceDocument4 pagesPHD Thesis Surface Plasmon Resonancevetepuwej1z3100% (1)
- TH - H BondingDocument5 pagesTH - H BondingDurga Prasad KalamNo ratings yet
- Acta Crystallographica Section D - 2018 - Kovalevskiy - Overview of Refinement Procedures Within REFMAC5 Utilizing DataDocument13 pagesActa Crystallographica Section D - 2018 - Kovalevskiy - Overview of Refinement Procedures Within REFMAC5 Utilizing DataAnna ŚciukNo ratings yet
- Nabuurs Et Al. Concepts Magn. Reson. 22A 90-105 2004Document16 pagesNabuurs Et Al. Concepts Magn. Reson. 22A 90-105 2004Rigel_TNo ratings yet
- X-Ray Crystallography 4Document5 pagesX-Ray Crystallography 4Miles NsgNo ratings yet
- Polym 12051053Document33 pagesPolym 12051053AynamawNo ratings yet
- Dissertation NMRDocument5 pagesDissertation NMRFindSomeoneToWriteMyCollegePaperUK100% (1)
- The Electron Density and Chemical Bonding in Organic Compounds by X-Ray DiffractionDocument58 pagesThe Electron Density and Chemical Bonding in Organic Compounds by X-Ray DiffractionJesus Isaac Castillo SanchezNo ratings yet
- Chemical Crystallography by Serial Femtosecond X-Ray DiffractionDocument16 pagesChemical Crystallography by Serial Femtosecond X-Ray DiffractionIgnetious MnisiNo ratings yet
- Swygenhoven Dislocations in Solids Vol 14-2Document42 pagesSwygenhoven Dislocations in Solids Vol 14-2huze_nedianNo ratings yet
- XRF InfoDocument4 pagesXRF InfoNicolucasNo ratings yet
- Chirality Supercoiling Assigned By: of DNA Scanning Force MicrosDocument4 pagesChirality Supercoiling Assigned By: of DNA Scanning Force MicrosUylrikkNo ratings yet
- Nature DislocationDocument6 pagesNature DislocationinekNo ratings yet
- ρ0 meson in the nucleusDocument10 pagesρ0 meson in the nucleusMani PillaiNo ratings yet
- TMP 51 BDocument7 pagesTMP 51 BFrontiersNo ratings yet
- Sample PreparationDocument13 pagesSample Preparationtanjiromanjiro20No ratings yet
- Maurer 2012Document5 pagesMaurer 2012asteriNo ratings yet
- Sali Structure 2005Document3 pagesSali Structure 2005hahahaNo ratings yet
- PCMT4401 X Ray and Electron Microscopy 7th Sem MMEDocument64 pagesPCMT4401 X Ray and Electron Microscopy 7th Sem MMEShibaji SahooNo ratings yet
- Material S. Presentation 1Document77 pagesMaterial S. Presentation 1Dagmawi HailuNo ratings yet
- Roughness PreviewDocument10 pagesRoughness PreviewFaisal AmirNo ratings yet
- Surface Plasmon Resonance ThesisDocument6 pagesSurface Plasmon Resonance Thesisbsna8p6k100% (2)
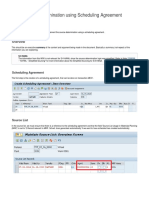
- MRP Source Determination Using Scheduling Agreement: PurposeDocument2 pagesMRP Source Determination Using Scheduling Agreement: PurposeBhanu TejNo ratings yet
- Practical 4 Servlet DemoDocument15 pagesPractical 4 Servlet DemoSoham JoshiNo ratings yet
- Bài tập ATHADocument2 pagesBài tập ATHALang Tuấn NguyênNo ratings yet
- CICS Training July 2009Document175 pagesCICS Training July 2009Ivan Petrucci100% (2)
- Beginning and Intermediate Algebra 6th Edition Lial Solutions ManualDocument90 pagesBeginning and Intermediate Algebra 6th Edition Lial Solutions ManualJamesWolfefsgr100% (46)
- Associate in Science Degree: Major RequirementsDocument5 pagesAssociate in Science Degree: Major Requirementsjoescribd55No ratings yet
- NC200U-B RF/DC Nanocluster SourceDocument25 pagesNC200U-B RF/DC Nanocluster SourceSantiago de la RivaNo ratings yet
- Telnet TNA670A00 150505 ENDocument2 pagesTelnet TNA670A00 150505 ENangicarNo ratings yet
- Engine - Repair - 1999-2000 Engine - Repair - AAA, AFP VR6Document19 pagesEngine - Repair - 1999-2000 Engine - Repair - AAA, AFP VR6Dany Pistiner100% (3)
- Roof PurlinsDocument20 pagesRoof PurlinsWegner KuchongNo ratings yet
- LR Mobile Diagnostic LogDocument97 pagesLR Mobile Diagnostic LogSalim AlsenaniNo ratings yet
- Treynor Black ModelDocument20 pagesTreynor Black ModelVaidyanathan RavichandranNo ratings yet
- AW139 Training Notes b2Document1,958 pagesAW139 Training Notes b2pbubs94% (34)
- GHTP Lehra MohabbatDocument23 pagesGHTP Lehra MohabbatDivanshu Garg40% (5)
- ES MT 0106 - REV1.30 - 24122019 - 6021282 DIAM 4100 Rev 1 30 - EN 2Document64 pagesES MT 0106 - REV1.30 - 24122019 - 6021282 DIAM 4100 Rev 1 30 - EN 2Paix AvousNo ratings yet
- DESIGN OF BEAM Bending Moment On A Beam Basic FormulaeDocument4 pagesDESIGN OF BEAM Bending Moment On A Beam Basic FormulaeGeorge KaraspNo ratings yet
- Metric Conversions and PracticeDocument5 pagesMetric Conversions and PracticekwilsonNo ratings yet
- 3de e (Bocr) ManualDocument6 pages3de e (Bocr) ManualNaveen Gupta100% (1)
- Excel Quickstart Guide From Beginner To Expert Excel Microsoft Office by William FischerDocument7 pagesExcel Quickstart Guide From Beginner To Expert Excel Microsoft Office by William FischerPruthvi Raja100% (1)
- Mathematics Research Paper SampleDocument7 pagesMathematics Research Paper Samplexwrcmecnd100% (1)
- Mo's AlgorithmDocument16 pagesMo's AlgorithmAmar MalikNo ratings yet
- How To Determine Sample Size, Determining Sample SizeDocument3 pagesHow To Determine Sample Size, Determining Sample SizeRiza AlfianNo ratings yet
- GB Catalog Update 2013-1 Inlay LRDocument296 pagesGB Catalog Update 2013-1 Inlay LRsaotinhyeu307783No ratings yet
- 3 Dispersion Skewness Kurtosis PDFDocument42 pages3 Dispersion Skewness Kurtosis PDFKelvin Kayode OlukojuNo ratings yet
- Click Here - : EnzymesDocument4 pagesClick Here - : EnzymesAmit GodaraNo ratings yet
- Kleyn Successful (2012) G1 MaterialDocument9 pagesKleyn Successful (2012) G1 MaterialcarlmarxNo ratings yet
- Physicochemical ProblemsDocument309 pagesPhysicochemical ProblemsNataniel LinaresNo ratings yet
- Comparative Evaluation of Initial Crestal Bone Loss Around Dental Implants Using Flapless or Flap Method: An in Vivo StudyDocument9 pagesComparative Evaluation of Initial Crestal Bone Loss Around Dental Implants Using Flapless or Flap Method: An in Vivo StudyIJAR JOURNALNo ratings yet
- Chemistry Textbook 10 - 12 PDFDocument86 pagesChemistry Textbook 10 - 12 PDFMusanta Simmon SimzNo ratings yet
- EP 8 CPD20TV8 Parts Manual 20190605 165931Document119 pagesEP 8 CPD20TV8 Parts Manual 20190605 165931Виталий ЧерновNo ratings yet