You might also like
- Solution Manual For Chemical Engineering ThermodynamicsDocument4 pagesSolution Manual For Chemical Engineering ThermodynamicsBayu Prayudi Wibowo8% (36)
- Analytical Chemistry Student Solutions Manual 6th Edition Gary D Christian Wiley PDFDocument86 pagesAnalytical Chemistry Student Solutions Manual 6th Edition Gary D Christian Wiley PDFJerico Ortega Santos100% (4)
- APA Tech Bulletin On FormadehydeDocument4 pagesAPA Tech Bulletin On Formadehydeca_alzuNo ratings yet
- 09-SAMSS-071 - (2016) Qualification Requirements For Inorganic Zinc Primer (APCS-17A) and (APCS-17B)Document9 pages09-SAMSS-071 - (2016) Qualification Requirements For Inorganic Zinc Primer (APCS-17A) and (APCS-17B)middlepermian100% (1)
- A Review of Silver Nanoparticles: Synthesis Methods, Properties and ApplicationsDocument8 pagesA Review of Silver Nanoparticles: Synthesis Methods, Properties and ApplicationsNaveenNo ratings yet
- Nanomaterials 13 00048Document14 pagesNanomaterials 13 00048safiaNo ratings yet
- Research ArticleDocument11 pagesResearch ArticleXyNo ratings yet
- Sans TitreDocument8 pagesSans TitreaminaNo ratings yet
- Arroya Ve 2020Document9 pagesArroya Ve 2020Karl ZeiessNo ratings yet
- Water 15 00384 v3Document24 pagesWater 15 00384 v3kamalugarba93No ratings yet
- Silver Modified Porous 3D Nitrogen-Doped Graphene Aerogel Highly Efficient Oxygen Reduction Electrocatalyst For ZN Air BatteryDocument38 pagesSilver Modified Porous 3D Nitrogen-Doped Graphene Aerogel Highly Efficient Oxygen Reduction Electrocatalyst For ZN Air Batterysiti khotijahNo ratings yet
- Facile Synthesis of Al Emg Co-Doped Zno Nanoparticles and Their High Hydrogen Sensing PerformancesDocument13 pagesFacile Synthesis of Al Emg Co-Doped Zno Nanoparticles and Their High Hydrogen Sensing PerformancesMohamed Ali DaymiNo ratings yet
- Dipti PaperDocument19 pagesDipti PaperShreyaNo ratings yet
- Electroplated Core-Shell NanowDocument9 pagesElectroplated Core-Shell NanowjulianNo ratings yet
- Synthesis and Deposition of Ag Nanoparticles by CoDocument9 pagesSynthesis and Deposition of Ag Nanoparticles by Comonikasharma1604No ratings yet
- Graphene Aerogel 1Document27 pagesGraphene Aerogel 1YATHISH M GNo ratings yet
- Influence of Inorganic Binder Composition On The SDocument9 pagesInfluence of Inorganic Binder Composition On The SLaura González SossaNo ratings yet
- Evaluation and Characterization of Reduced Graphene Oxide Nanosheets As Anode Materials For Lithium-Ion BatteriesDocument12 pagesEvaluation and Characterization of Reduced Graphene Oxide Nanosheets As Anode Materials For Lithium-Ion BatteriesRisa Rahmawati PurliantoroNo ratings yet
- Discover Nano: Doped MnoDocument16 pagesDiscover Nano: Doped MnoMarta FrançaNo ratings yet
- Synergic Effect of Plasmonic Gold NanoparticlesDocument10 pagesSynergic Effect of Plasmonic Gold NanoparticlesAdil KhanNo ratings yet
- Accepted Manuscript: Computational & Theoretical ChemistryDocument46 pagesAccepted Manuscript: Computational & Theoretical ChemistrySalah AlchimistNo ratings yet
- 1 s2.0 S266608652200087X MainDocument9 pages1 s2.0 S266608652200087X Mainowais khattakNo ratings yet
- 2017 Novel Concept For Fabricating A Flexible Transparent Electrode (The JOURNAL of PHYSICAL CHEMISTRY C)Document7 pages2017 Novel Concept For Fabricating A Flexible Transparent Electrode (The JOURNAL of PHYSICAL CHEMISTRY C)최요민No ratings yet
- Biomass To BiofuelDocument9 pagesBiomass To BiofuelPallab RoyNo ratings yet
- 3 - Intermetallic Compounds of Ni and Ga As Catalysts For The Synthesis of MethanolDocument12 pages3 - Intermetallic Compounds of Ni and Ga As Catalysts For The Synthesis of Methanoltunganh1110No ratings yet
- Hydrothermal ProcessDocument28 pagesHydrothermal Processdeepakrm74No ratings yet
- Nanocomposite 2023 N3 To NH3Document18 pagesNanocomposite 2023 N3 To NH3Eva AberaNo ratings yet
- The Optimization of Effective Parameters For Electrodeposition of Reduced Graphene Oxide Through Taguchi Method To Evaluate The Charge TransferDocument8 pagesThe Optimization of Effective Parameters For Electrodeposition of Reduced Graphene Oxide Through Taguchi Method To Evaluate The Charge TransferYosseline Vargas MezaNo ratings yet
- Dye-Sensitized Solar Cells: Fundamentals and Current Status: Nanoreview Open AccessDocument46 pagesDye-Sensitized Solar Cells: Fundamentals and Current Status: Nanoreview Open AccessKrishna PatelNo ratings yet
- Synthesis of Ammonia Gas Sensor Based On Multiwalled Carbon Nanotube and Schiff Base PolymerDocument43 pagesSynthesis of Ammonia Gas Sensor Based On Multiwalled Carbon Nanotube and Schiff Base Polymermohammed mahadi.aNo ratings yet
- Ag ZnODocument10 pagesAg ZnORobeul AwalNo ratings yet
- RSC Books: DefiningDocument9 pagesRSC Books: DefiningFrontiersNo ratings yet
- Graphite PowderDocument7 pagesGraphite Powderadam_723172810No ratings yet
- J Jallcom 2015 09 241Document8 pagesJ Jallcom 2015 09 241THƯ NGUYỄN MINHNo ratings yet
- Design and Development of A Novel CellulDocument9 pagesDesign and Development of A Novel Celluldevi asrianiNo ratings yet
- Carbon: Boya Kuang, Weili Song, Mingqiang Ning, Jingbo Li, Zhengjing Zhao, Deyu Guo, Maosheng Cao, Haibo JinDocument9 pagesCarbon: Boya Kuang, Weili Song, Mingqiang Ning, Jingbo Li, Zhengjing Zhao, Deyu Guo, Maosheng Cao, Haibo JinwidyaNo ratings yet
- Accepted Manuscript: Applied Catalysis B: EnvironmentalDocument46 pagesAccepted Manuscript: Applied Catalysis B: EnvironmentalJulian Ruiz MejiaNo ratings yet
- Silver/Polyaniline Composite Nanotubes: One-Step Synthesis and Electrocatalytic Activity For Neurotransmitter DopamineDocument7 pagesSilver/Polyaniline Composite Nanotubes: One-Step Synthesis and Electrocatalytic Activity For Neurotransmitter DopamineGil RochaNo ratings yet
- Non-Metal Doping of Transition Metal Oxides For Visible-Light PhotocatalysisDocument25 pagesNon-Metal Doping of Transition Metal Oxides For Visible-Light Photocatalysispetru apopeiNo ratings yet
- 1 s2.0 S027288422401157X MainDocument32 pages1 s2.0 S027288422401157X MainPioneros de lo NanoNo ratings yet
- Macro-Microporous Carbon With A Three-Dimensional Channel From Waste Sun Flower Seed ShellDocument9 pagesMacro-Microporous Carbon With A Three-Dimensional Channel From Waste Sun Flower Seed ShellMuhamad SuharNo ratings yet
- Sno 2Document32 pagesSno 2Muhammad UsmanNo ratings yet
- Han2016 PDFDocument34 pagesHan2016 PDFTiken TamuliNo ratings yet
- Antibacterial and Photocatalytic Properties of Ag/Tio /zno Nano - Owers Prepared by Facile One-Pot Hydrothermal ProcessDocument8 pagesAntibacterial and Photocatalytic Properties of Ag/Tio /zno Nano - Owers Prepared by Facile One-Pot Hydrothermal ProcessDaniel ReyesNo ratings yet
- Catalysts: Selective Catalytic Reduction of NODocument4 pagesCatalysts: Selective Catalytic Reduction of NOGodfrey Eric MuendoNo ratings yet
- Tunable ConductivityDocument10 pagesTunable ConductivityChristhy Vanessa Ruiz MadroñeroNo ratings yet
- Fabrication of Nitrogen-Doped Porous Electrically Conductive Carbon Aerogel From Waste Cabbage For Supercapacitors and Oil Water SeparationDocument11 pagesFabrication of Nitrogen-Doped Porous Electrically Conductive Carbon Aerogel From Waste Cabbage For Supercapacitors and Oil Water SeparationAnh DuyNo ratings yet
- Advances of Ag, Cu, and Ag-Cu Alloy Nanoparticles SynthesizedDocument29 pagesAdvances of Ag, Cu, and Ag-Cu Alloy Nanoparticles SynthesizedThamirisAgmNo ratings yet
- The Role of Graphene OxideDocument17 pagesThe Role of Graphene Oxidelibrary inspenNo ratings yet
- Accepted Manuscript: Colloids and Surfaces A: Physicochem. Eng. AspectsDocument36 pagesAccepted Manuscript: Colloids and Surfaces A: Physicochem. Eng. AspectsNguyen Huu HieuNo ratings yet
- Artigo BateriaDocument8 pagesArtigo BateriaJoanaNo ratings yet
- Dielectric Properties of MgOEpoxy Nanocomposites IDocument6 pagesDielectric Properties of MgOEpoxy Nanocomposites IZoalfokkar Al-ObadNo ratings yet
- Chandramika BoraDocument7 pagesChandramika BoraMahalingam SNo ratings yet
- Chemical Engineering Journal: 2 Paula Navascu Es, Jose Cotrino, Agustín R. Gonz Alez-Elipe, Ana G Omez-RamírezDocument10 pagesChemical Engineering Journal: 2 Paula Navascu Es, Jose Cotrino, Agustín R. Gonz Alez-Elipe, Ana G Omez-RamírezMD Redwan IslamNo ratings yet
- UTMFR Grant - RevDocument5 pagesUTMFR Grant - RevNur Isarah Che RaimiNo ratings yet
- Agulhon 2012Document6 pagesAgulhon 2012brouuorbNo ratings yet
- Moustafa Et Al. - 2020 - Titanium Dioxide-Decorated RGO As An Effective EleDocument25 pagesMoustafa Et Al. - 2020 - Titanium Dioxide-Decorated RGO As An Effective EleHuỳnh Tuấn KiệtNo ratings yet
- Applied Surface Science: M.A. Gondal, Q.A. Drmosh, Z.H. Yamani, T.A. SalehDocument7 pagesApplied Surface Science: M.A. Gondal, Q.A. Drmosh, Z.H. Yamani, T.A. Salehsgc17No ratings yet
- Vidhu2015 - Sno2 AntibacteriaDocument8 pagesVidhu2015 - Sno2 AntibacteriaBagavananth RajNo ratings yet
- 1 s2.0 S0021979716307147 MainDocument11 pages1 s2.0 S0021979716307147 MainhrithvinNo ratings yet
- 1 s2.0 S1872206721639049 MainDocument12 pages1 s2.0 S1872206721639049 MainIsraelPala-RosasNo ratings yet
- Final Report SurfaceDocument2 pagesFinal Report SurfacenandaNo ratings yet
- Nanocrystalline Materials: Their Synthesis-Structure-Property Relationships and ApplicationsFrom EverandNanocrystalline Materials: Their Synthesis-Structure-Property Relationships and ApplicationsNo ratings yet
- Heterogeneous Nanocomposite-Photocatalysis for Water PurificationFrom EverandHeterogeneous Nanocomposite-Photocatalysis for Water PurificationNo ratings yet
- Flammability TestsDocument15 pagesFlammability TestsA MahmoodNo ratings yet
- Influence Venner Formaldehyde EmissionDocument4 pagesInfluence Venner Formaldehyde Emissionca_alzuNo ratings yet
- Introduction and Course Description: Introduction To The SubjectDocument5 pagesIntroduction and Course Description: Introduction To The SubjectskbeheraNo ratings yet
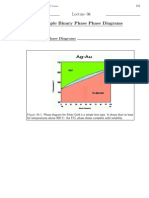
- Example Phase Diagrams: MIT 3.00 Fall 2002 ° W.C CarterDocument4 pagesExample Phase Diagrams: MIT 3.00 Fall 2002 ° W.C Carterca_alzuNo ratings yet
- PEG PNIPAM TermoresponsiveDocument5 pagesPEG PNIPAM Termoresponsiveca_alzuNo ratings yet
- Effective Science Talks: Celia M. ElliottDocument40 pagesEffective Science Talks: Celia M. ElliottHi_LevNo ratings yet
- Spotlight On Ionic LiquidsDocument10 pagesSpotlight On Ionic Liquidsca_alzuNo ratings yet
- Whisky TourDocument5 pagesWhisky Tourca_alzuNo ratings yet
- BS National Structural Concrete Specification For Building ConstructionDocument132 pagesBS National Structural Concrete Specification For Building ConstructionAmie D MalobagoNo ratings yet
- BS National Structural Concrete Specification For Building ConstructionDocument132 pagesBS National Structural Concrete Specification For Building ConstructionAmie D MalobagoNo ratings yet
- BS National Structural Concrete Specification For Building ConstructionDocument132 pagesBS National Structural Concrete Specification For Building ConstructionAmie D MalobagoNo ratings yet
- Smart Polymers and Their Applications As Biomaterials: M.R.Aguilar, C. Elvira, A. Gallardo, B. Vázquez, and J.S. RománDocument27 pagesSmart Polymers and Their Applications As Biomaterials: M.R.Aguilar, C. Elvira, A. Gallardo, B. Vázquez, and J.S. RománAbdul HakimNo ratings yet
- Effective Science Talks: Celia M. ElliottDocument40 pagesEffective Science Talks: Celia M. ElliottHi_LevNo ratings yet
- Materials 07 00805Document71 pagesMaterials 07 00805ca_alzuNo ratings yet
- Modeling and Analysis of The Compatibility of Polystyrene/poly (Methyl Methacrylate) Blends With Four Inducing EffectsDocument13 pagesModeling and Analysis of The Compatibility of Polystyrene/poly (Methyl Methacrylate) Blends With Four Inducing Effectsca_alzuNo ratings yet
- Preparation, Structure and Morphology of Polymer Supports: David C. SherringtonDocument12 pagesPreparation, Structure and Morphology of Polymer Supports: David C. Sherringtonkishorkumarn8212No ratings yet
- Predominance Zone Diagrams in SolutionDocument7 pagesPredominance Zone Diagrams in Solutionca_alzuNo ratings yet
- Fluctuation-Dissipation Theorem (FDT) : 1 Classical MechanicsDocument6 pagesFluctuation-Dissipation Theorem (FDT) : 1 Classical Mechanicsca_alzuNo ratings yet
- Modeling The Rheology of Polymer Solutions by Dissipative Particle DynamicsDocument6 pagesModeling The Rheology of Polymer Solutions by Dissipative Particle Dynamicsca_alzuNo ratings yet
- FDT2 PDFDocument148 pagesFDT2 PDFca_alzuNo ratings yet
- An Investigation Into The Morphologies of PolystyDocument123 pagesAn Investigation Into The Morphologies of Polystyca_alzuNo ratings yet
- Grade 6 ScienceDocument2 pagesGrade 6 ScienceThao Trinh0% (1)
- AFM CFD Simulation ThesisDocument129 pagesAFM CFD Simulation ThesisRanveer AubeeluckNo ratings yet
- Sanyo Alarm Codes PDFDocument6 pagesSanyo Alarm Codes PDFomarNo ratings yet
- Tutorial 17 Rapid DrawdownDocument17 pagesTutorial 17 Rapid DrawdownJustoArteagaHuacchaNo ratings yet
- GP2 Electric-FieldDocument28 pagesGP2 Electric-FieldChristine Karel Marcelino0% (1)
- Combustion Chemstry1Document6 pagesCombustion Chemstry1Dr Mohammad AlzoubyNo ratings yet
- Separation Assignment MembraneDocument4 pagesSeparation Assignment MembraneQilah KamarudinNo ratings yet
- P436 Lect 10p5Document15 pagesP436 Lect 10p5Zhenhua HuangNo ratings yet
- Dielectric Properties of CoalDocument11 pagesDielectric Properties of CoalEsther BarrachinaNo ratings yet
- T7 - Mid-Semister Revisit and SterilizationDocument4 pagesT7 - Mid-Semister Revisit and SterilizationFishNo ratings yet
- Waterstops PDFDocument26 pagesWaterstops PDFjmusopoleNo ratings yet
- Remember TheseDocument34 pagesRemember Thesem_alodat6144No ratings yet
- Homogeneous and Structured PCD-WC-Co Materials For DrillingDocument9 pagesHomogeneous and Structured PCD-WC-Co Materials For Drillingdan_cunningham_15No ratings yet
- Design Os Sewers (Blue Book)Document388 pagesDesign Os Sewers (Blue Book)savanotrebor100% (1)
- Ballastguard Cip Cleaner 25 LTRDocument2 pagesBallastguard Cip Cleaner 25 LTRsijinjoyNo ratings yet
- Music TherapyDocument13 pagesMusic TherapyXavier KiranNo ratings yet
- 06 05 16 Heko Bucket ElevatorDocument36 pages06 05 16 Heko Bucket ElevatorLeoncio Alexander Maza InfantesNo ratings yet
- Chap 1 Thermodynamics Exercise PDFDocument25 pagesChap 1 Thermodynamics Exercise PDFRanveer GautamNo ratings yet
- EE Catalogue PDFDocument27 pagesEE Catalogue PDFssnNo ratings yet
- Paper BioDocument17 pagesPaper BioI-hana D'yanaNo ratings yet
- Rotogravure Printing Learn MoreDocument15 pagesRotogravure Printing Learn Moreumangashling100% (1)
- M 3031 (2013-06)Document16 pagesM 3031 (2013-06)Hatada FelipeNo ratings yet
- Zaikov CVDocument3 pagesZaikov CVArif SantosoNo ratings yet
- Aipmt 1999Document16 pagesAipmt 1999sumit kumarNo ratings yet
- Sabkha Trafficability, JubailDocument163 pagesSabkha Trafficability, JubailAdly Al-SaafinNo ratings yet
- Homework 7-1 Modern ChemistryDocument6 pagesHomework 7-1 Modern Chemistryafnapvbseurfgy100% (1)
- 1989 - Rapid Extraction of Bacterial Genomic DNA With Guanidium ThiocyanateDocument6 pages1989 - Rapid Extraction of Bacterial Genomic DNA With Guanidium ThiocyanateramarquezoNo ratings yet
- 985 22647-R02 Installation Manual ORCA Offshore Part1Document21 pages985 22647-R02 Installation Manual ORCA Offshore Part1dumpuu100% (1)