You might also like
- Justine's Memoirs On Actual Freedom (Part - I)Document61 pagesJustine's Memoirs On Actual Freedom (Part - I)JustineNo ratings yet
- IMO Class 3 Sample Paper 2017 18Document3 pagesIMO Class 3 Sample Paper 2017 18sunil KumarNo ratings yet
- PML Manual PDFDocument178 pagesPML Manual PDFChandrashekar R100% (1)
- Data Encryption DecryptionDocument60 pagesData Encryption DecryptionMohit Sharma100% (2)
- SP Racing F3 FC Board(Deluxe) internal OSD Specification and SetupDocument5 pagesSP Racing F3 FC Board(Deluxe) internal OSD Specification and SetupibyselfNo ratings yet
- CH 02Document20 pagesCH 02Pauline Nguyen100% (1)
- Dehydrogenation by Heterogeneous CatalystsDocument52 pagesDehydrogenation by Heterogeneous CatalystsSankar SasmalNo ratings yet
- TCS NotesDocument10 pagesTCS Notesdhana sethupathyNo ratings yet
- Hydrogenation of Ethylene On Cu CatalystDocument7 pagesHydrogenation of Ethylene On Cu CatalystHillman WiraNo ratings yet
- Austrian 25Document13 pagesAustrian 25Vo Tung LamNo ratings yet
- Cataltic Applications of Metal CarbonylsDocument8 pagesCataltic Applications of Metal Carbonylssaud100% (4)
- Reaksi Kopling Merupakan Reaksi Penggabungan Rantai KarbonDocument9 pagesReaksi Kopling Merupakan Reaksi Penggabungan Rantai KarbonfikarisvitaNo ratings yet
- Aldol-Type Coupling of Aldehydes With Ethyl Diazoacetate Catalyzed by Supported Ionic Liquid PDFDocument4 pagesAldol-Type Coupling of Aldehydes With Ethyl Diazoacetate Catalyzed by Supported Ionic Liquid PDFsarabinduroyNo ratings yet
- Lim 20071Document5 pagesLim 20071gregorbanalt1No ratings yet
- Gmewue: International Edition in EnglishDocument14 pagesGmewue: International Edition in English560712No ratings yet
- Epoxidation of Alkenes With Hydrogen PeroxideDocument7 pagesEpoxidation of Alkenes With Hydrogen PeroxidechidambaramrNo ratings yet
- Tetrahedron Letters: Payal Malik, Debashis ChakrabortyDocument3 pagesTetrahedron Letters: Payal Malik, Debashis ChakrabortyBagusChandraMahardhikaNo ratings yet
- Esterification Process To Synthesize Isopropyl Chloroacetate Catalyzed by Lanthanum Dodecyl SulfateDocument6 pagesEsterification Process To Synthesize Isopropyl Chloroacetate Catalyzed by Lanthanum Dodecyl SulfateVinay JainNo ratings yet
- 10 1016@j Apcata 2011 04 046Document10 pages10 1016@j Apcata 2011 04 046farah al-sudaniNo ratings yet
- tmpF0F1 TMPDocument11 pagestmpF0F1 TMPFrontiersNo ratings yet
- Selective Extraction of Neutral Nitrogen Compounds Found in Diesel Feed byDocument8 pagesSelective Extraction of Neutral Nitrogen Compounds Found in Diesel Feed byJohnSmithNo ratings yet
- Dehydrogenation by Heterogeneous CatalystsDocument52 pagesDehydrogenation by Heterogeneous CatalystsNur GeehanNo ratings yet
- Mild Aerobic Oxidative Deoximation using Sodium Nitrite CatalystDocument4 pagesMild Aerobic Oxidative Deoximation using Sodium Nitrite CatalystCláudio SerafimNo ratings yet
- Jaime de La Sota PDFDocument6 pagesJaime de La Sota PDFbryan10032013No ratings yet
- Tetrahedron Letters 52 (2011) 5107-5109Document3 pagesTetrahedron Letters 52 (2011) 5107-5109rajesh_tammana3550No ratings yet
- HDO-aducto-Catal Today 2016Document8 pagesHDO-aducto-Catal Today 2016cligcodiNo ratings yet
- New Montmorillonite Silylpropylethylenediamine Palladium (II) Complex in Oxidation of Terminal OlefinsDocument6 pagesNew Montmorillonite Silylpropylethylenediamine Palladium (II) Complex in Oxidation of Terminal OlefinsChamula K MasNo ratings yet
- Enantioselective Cyclopropenation of Alkynes With Acceptor/Acceptor-Substituted Diazo Reagents Via Co (II) - Based Metalloradical CatalysisDocument4 pagesEnantioselective Cyclopropenation of Alkynes With Acceptor/Acceptor-Substituted Diazo Reagents Via Co (II) - Based Metalloradical CatalysiskyawmoetunNo ratings yet
- Homogeneous CatalystDocument52 pagesHomogeneous CatalystParom WaikasikarnNo ratings yet
- Hydrogen From SMR 2Document2 pagesHydrogen From SMR 2Pramanshu RajputNo ratings yet
- 10 1016@j Colsurfa 2017 04 046Document31 pages10 1016@j Colsurfa 2017 04 046alfina ameliaNo ratings yet
- Basic Yellow Conversion Ni - MgAlODocument6 pagesBasic Yellow Conversion Ni - MgAlONAJAT EL KHAOUANo ratings yet
- Morales2015 PDFDocument4 pagesMorales2015 PDFAlejandraNo ratings yet
- Chem. Rev., 1974, 74 (5), PP 567-580Document14 pagesChem. Rev., 1974, 74 (5), PP 567-580sibbanac acidniNo ratings yet
- Surface Chemistry: Adsorption From SolutionsDocument27 pagesSurface Chemistry: Adsorption From SolutionsSrijan GoyalNo ratings yet
- Thermal Modification Effect On Supported Cu-BasedDocument17 pagesThermal Modification Effect On Supported Cu-Basedsyarif hidayatNo ratings yet
- Effects of Solvent Polarity On The Hydrogenation of Xylose: Jyri-Pekka Mikkola, Tapio Salmi and Rainer Sjo HolmDocument11 pagesEffects of Solvent Polarity On The Hydrogenation of Xylose: Jyri-Pekka Mikkola, Tapio Salmi and Rainer Sjo HolmEdgar Fernando Jerez GarciaNo ratings yet
- Ionic Liquids Perspectives For Organic and Catalytic Reactions, Olivier y MagnaDocument19 pagesIonic Liquids Perspectives For Organic and Catalytic Reactions, Olivier y MagnaIvan Jose Acosta MoralesNo ratings yet
- Jave PDFDocument10 pagesJave PDFjaveria tahirNo ratings yet
- JPPS 0209 Zhen GalleyDocument8 pagesJPPS 0209 Zhen GalleyGregorio ValeroNo ratings yet
- Aldehid Dan KetonDocument5 pagesAldehid Dan KetonVicha FatanahNo ratings yet
- Ijhe 29 5 2004Document6 pagesIjhe 29 5 2004VolodymyrNo ratings yet
- HDO Nickel CatalisisDocument8 pagesHDO Nickel CatalisisPAULA ALARCON CAMPOSNo ratings yet
- Production of Renewable Hydrogen by Aqueous-Phase Reforming of Glycerol OverDocument7 pagesProduction of Renewable Hydrogen by Aqueous-Phase Reforming of Glycerol OverMahdy HajienayatiNo ratings yet
- Kuk Lin 2016Document6 pagesKuk Lin 2016Valeria AlarcónNo ratings yet
- Abcd. Proiect AdamDocument14 pagesAbcd. Proiect AdamSfiriac LauraNo ratings yet
- Route of The Catalytic Oxidation of Phenol in Aqueous PhaseDocument17 pagesRoute of The Catalytic Oxidation of Phenol in Aqueous Phaseedgarpinzon21No ratings yet
- Oxidation of Glycerol To Lactic Acid by Gold OnDocument21 pagesOxidation of Glycerol To Lactic Acid by Gold OnlarguedasNo ratings yet
- Presentation by GopalDocument19 pagesPresentation by GopalSherlock BakshiNo ratings yet
- Benz AldehydeDocument5 pagesBenz AldehydeRoni BaroesNo ratings yet
- Islam2011 PDFDocument8 pagesIslam2011 PDFOussamaNeharNo ratings yet
- Hydrolisis of EthanodiolDocument6 pagesHydrolisis of Ethanodiolchama_gozNo ratings yet
- ApplCat B-2014-SalvadorDocument9 pagesApplCat B-2014-SalvadorcligcodiNo ratings yet
- Karski 2003Document5 pagesKarski 2003farah al-sudaniNo ratings yet
- Zhiyi Cui Natcat 2018Document7 pagesZhiyi Cui Natcat 2018CHEMICALNo ratings yet
- Carbohydrate-derived carbon cryogels activate persulfateDocument42 pagesCarbohydrate-derived carbon cryogels activate persulfateEcNo ratings yet
- 1 s2.0 S0926337322008992 MainDocument11 pages1 s2.0 S0926337322008992 MainAlbanny PerezNo ratings yet
- Paper 3 PDFDocument8 pagesPaper 3 PDFJimmy NelsonNo ratings yet
- Kelompok 1Document7 pagesKelompok 1Roby SudarmanNo ratings yet
- 1 s2.0 S0926860X08000963 MainDocument9 pages1 s2.0 S0926860X08000963 Mainpetru apopeiNo ratings yet
- Biocatalytic Oxidation of Benzyl Alcohol via Hydrogen TransferDocument5 pagesBiocatalytic Oxidation of Benzyl Alcohol via Hydrogen TransferMario Benito PeinadoNo ratings yet
- 1 s2.0 S0040403906014444 Main PDFDocument3 pages1 s2.0 S0040403906014444 Main PDFPrasanth BitlaNo ratings yet
- Production of Hydrogen by Steam Reforming of Ethanol Over A Ni/Zno CatalystDocument6 pagesProduction of Hydrogen by Steam Reforming of Ethanol Over A Ni/Zno Catalystpetro121No ratings yet
- Kinetics and Related Engineering Aspects of Catalytic Liquid-Phase Oxidation of P-Xylene To Terephthalic AcidDocument17 pagesKinetics and Related Engineering Aspects of Catalytic Liquid-Phase Oxidation of P-Xylene To Terephthalic AcidAbdullah JavedNo ratings yet
- Coupling Reaction - Wikipedia, The Free EncyclopediaDocument5 pagesCoupling Reaction - Wikipedia, The Free EncyclopediakavitakunduNo ratings yet
- Transition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesFrom EverandTransition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesNo ratings yet
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesFrom EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesNo ratings yet
- Tips On Getting Into Grad SchoolDocument16 pagesTips On Getting Into Grad SchoolasdhaksjhNo ratings yet
- Cerium ChlorideDocument4 pagesCerium ChlorideVo Tung LamNo ratings yet
- Symmetry Analysis For Vibrational Modes of BRF Using Group TheoryDocument13 pagesSymmetry Analysis For Vibrational Modes of BRF Using Group TheoryVo Tung LamNo ratings yet
- Results:: 2 4 (Aq) (Aq) (Aq) 2(s) 2 2 4 (Aq) 4 (Aq) 2 4(s) 3 2 4 3 2 2 2 4 (Aq) 2 2 (Aq) 2 2 4 (Aq)Document2 pagesResults:: 2 4 (Aq) (Aq) (Aq) 2(s) 2 2 4 (Aq) 4 (Aq) 2 4(s) 3 2 4 3 2 2 2 4 (Aq) 2 2 (Aq) 2 2 4 (Aq)Vo Tung LamNo ratings yet
- Craig JChemEdDocument7 pagesCraig JChemEdVo Tung LamNo ratings yet
- Should Congress Allow The Buying of Human Organs YES and NODocument4 pagesShould Congress Allow The Buying of Human Organs YES and NOVo Tung LamNo ratings yet
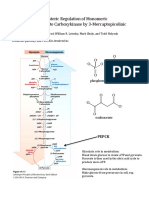
- Hints Sheet For PEPCK Kinetics PaperDocument2 pagesHints Sheet For PEPCK Kinetics PaperVo Tung LamNo ratings yet
- Photocrosslinking and Click Chemistry Enable The Specific Detection of Proteins Interacting With Phospholipids at The Membrane InterfaceDocument12 pagesPhotocrosslinking and Click Chemistry Enable The Specific Detection of Proteins Interacting With Phospholipids at The Membrane InterfaceVo Tung LamNo ratings yet
- Srs Drugs Cult Syllabus Win 2014Document7 pagesSrs Drugs Cult Syllabus Win 2014Vo Tung LamNo ratings yet
- Lam Vo Bus Ticket ConfirmationDocument2 pagesLam Vo Bus Ticket ConfirmationVo Tung LamNo ratings yet
- Fall Term 2013 Scheduling Instructions FOR UP-TO-DATE Information Visit The Web atDocument23 pagesFall Term 2013 Scheduling Instructions FOR UP-TO-DATE Information Visit The Web atVo Tung LamNo ratings yet
- TobermoryDocument1 pageTobermoryVo Tung LamNo ratings yet
- Writing Ink Identification: Standard Guide ForDocument5 pagesWriting Ink Identification: Standard Guide ForEric GozzerNo ratings yet
- A+ Guide to Managing Your PC Hardware & SoftwareDocument34 pagesA+ Guide to Managing Your PC Hardware & Software2AdvanceNo ratings yet
- NetAct Plan Editor 4.9-4 CNDocument4 pagesNetAct Plan Editor 4.9-4 CNAshraf JarjeesNo ratings yet
- Methodology of Education Research MCQSDocument12 pagesMethodology of Education Research MCQSRAFIULLAHNo ratings yet
- What Is Family Life CycleDocument7 pagesWhat Is Family Life Cycleapi-414688913No ratings yet
- Distribution Transformer Manufacturing Process ManualDocument29 pagesDistribution Transformer Manufacturing Process ManualAnonymous Jf6X8nNo ratings yet
- ExportDocument361 pagesExportStefanNo ratings yet
- Muhamad Azamudin ResumeDocument1 pageMuhamad Azamudin ResumeMuhamad AzamudinNo ratings yet
- Overqualified Cover LetterDocument4 pagesOverqualified Cover Letteroyutlormd100% (1)
- 11.servlet WrappersDocument14 pages11.servlet WrapperskasimNo ratings yet
- Parallels Between Schopenhauer and WittgensteinDocument1 pageParallels Between Schopenhauer and WittgensteinkwsxNo ratings yet
- 11-1203 Syed Hussain HaiderDocument16 pages11-1203 Syed Hussain HaiderSalman Nisar BhattiNo ratings yet
- Lattitude and Longitude PDF ProblemDocument2 pagesLattitude and Longitude PDF ProblemSatyendranath KarNo ratings yet
- Basculas Con MIGO SAPDocument3 pagesBasculas Con MIGO SAPmizraimNo ratings yet
- Beno K Pradekso - Solusi247 - In40aiDocument36 pagesBeno K Pradekso - Solusi247 - In40aiMuhammad HattaNo ratings yet
- Pork Carcass ChillingDocument6 pagesPork Carcass ChillingDumitru PodgorneakNo ratings yet
- Final QuestionDocument5 pagesFinal QuestionrahulNo ratings yet
- Costing 1 PDFDocument8 pagesCosting 1 PDFSamyNo ratings yet
- LZW Fundamentals: Lempel Ziv 1977 1978 Terry Welch's 1978 Algorithm 1984Document9 pagesLZW Fundamentals: Lempel Ziv 1977 1978 Terry Welch's 1978 Algorithm 1984Vishal PatilNo ratings yet
- NAHRIM - Institut Penyelidikan Hidraulik Kebangsaan Malaysia - Rainwater Harvesting SystemDocument4 pagesNAHRIM - Institut Penyelidikan Hidraulik Kebangsaan Malaysia - Rainwater Harvesting SystemAnonymous e1j2F5Ge0No ratings yet
- Osmotic Power Potential Questions and AnswersDocument12 pagesOsmotic Power Potential Questions and AnswersAishwar RavichandranNo ratings yet
- Integrating Information About The Cost of Carbon Through Activity Based Costing 2012 Journal of Cleaner ProductionDocument10 pagesIntegrating Information About The Cost of Carbon Through Activity Based Costing 2012 Journal of Cleaner ProductionLilian BrodescoNo ratings yet
- FormworksDocument94 pagesFormworksLouie Zavalla LeyvaNo ratings yet