You might also like
- CEFA8 D 01Document56 pagesCEFA8 D 01idhem1110No ratings yet
- Open Source Proteomics SoftwareDocument49 pagesOpen Source Proteomics Softwareken tsaiNo ratings yet
- Skyline Absolute QuantificationDocument15 pagesSkyline Absolute QuantificationShahinuzzamanAdaNo ratings yet
- Skyline Absolute QuantificationDocument17 pagesSkyline Absolute QuantificationShahinuzzamanAdaNo ratings yet
- Protein Mass Fingerprinting Protein Profiling Protein Assay: BIO105 / AC1Document26 pagesProtein Mass Fingerprinting Protein Profiling Protein Assay: BIO105 / AC1Christeliza Flores RaymundoNo ratings yet
- Artikel SIAM EngDocument8 pagesArtikel SIAM EngeferrarijrNo ratings yet
- 2012 LCGC SpecCTMS0512Document6 pages2012 LCGC SpecCTMS0512MLGPNo ratings yet
- 1265 FullDocument8 pages1265 FullArvind KumarNo ratings yet
- Principles and Applications of ProteomicsDocument39 pagesPrinciples and Applications of ProteomicsbiokanagasundarNo ratings yet
- Analyze The Data: Protein PurificationDocument3 pagesAnalyze The Data: Protein PurificationQuan ThieuNo ratings yet
- Bioinformatics LAb ReportDocument7 pagesBioinformatics LAb ReportBriana Halbert100% (3)
- Early History of Microbiological TechniqueDocument5 pagesEarly History of Microbiological TechniqueFadare Shadrach ONo ratings yet
- Bdox D Met Dioxins Furans Brominated-DioxinsDocument50 pagesBdox D Met Dioxins Furans Brominated-Dioxinssabrina amaralNo ratings yet
- Thesis Protein PurificationDocument6 pagesThesis Protein Purificationauroratuckernewyork100% (2)
- Food CompendiumDocument1,953 pagesFood CompendiumEddie Mendoza100% (1)
- PEAKS CompareSoftwareDocument1 pagePEAKS CompareSoftwareguruveer.singhNo ratings yet
- HW 13Document6 pagesHW 13David M RodgersNo ratings yet
- Proteomics Workshop SessionsDocument13 pagesProteomics Workshop SessionsFred SteeleNo ratings yet
- Agilent TXT File Pre-Processing Engine PDFDocument42 pagesAgilent TXT File Pre-Processing Engine PDFscjofyWFawlroa2r06YFVabfbajNo ratings yet
- Protein Purification Lecture (Ostap)Document14 pagesProtein Purification Lecture (Ostap)Gabriel OtteNo ratings yet
- Manual For: LFQ-AnalystDocument19 pagesManual For: LFQ-AnalystSergio CarreñoNo ratings yet
- 10.1016@j.mex.2020.100919experimental and Statistical Protocol For The Validation MethodsDocument8 pages10.1016@j.mex.2020.100919experimental and Statistical Protocol For The Validation MethodsAngel GarciaNo ratings yet
- Bioinformatics Seminar3rdOct18Document25 pagesBioinformatics Seminar3rdOct18subhasree majumderNo ratings yet
- Quantitative RT-PCR Protocol (Sybr Green I)Document8 pagesQuantitative RT-PCR Protocol (Sybr Green I)u77No ratings yet
- DNA Extraction: Qualitative Estimation of Genomic DNADocument32 pagesDNA Extraction: Qualitative Estimation of Genomic DNAPAWANKUMAR S. K.No ratings yet
- Name: TF Name:: LS1a Fall 2014 Lab 3 In-Lab ActivityDocument4 pagesName: TF Name:: LS1a Fall 2014 Lab 3 In-Lab ActivityThysianNo ratings yet
- Nitrogen-Containing Steroids As Potential Anticancer AgentsDocument31 pagesNitrogen-Containing Steroids As Potential Anticancer AgentsSamuelNo ratings yet
- Ismb99 008Document8 pagesIsmb99 008Orion OriNo ratings yet
- Module in TicsDocument20 pagesModule in TicsEmyadNo ratings yet
- Manual de Laboratorio Cimmyt PDFDocument102 pagesManual de Laboratorio Cimmyt PDFZettNo ratings yet
- Whatman Price ListDocument44 pagesWhatman Price ListSivananth MurugesanNo ratings yet
- Mass Spectrometry in ProteomicsDocument38 pagesMass Spectrometry in ProteomicsDawlat SalamaNo ratings yet
- Restriction Digestion and Analertysis of Lambda DNA Student ManualDocument24 pagesRestriction Digestion and Analertysis of Lambda DNA Student ManualSaswat DasNo ratings yet
- Computer Method 13c NMRDocument10 pagesComputer Method 13c NMRcarloschaverriNo ratings yet
- Identification of FungiDocument5 pagesIdentification of FungiYến NguyễnNo ratings yet
- Making Sense of The LCMS Data Differences - David WeilDocument65 pagesMaking Sense of The LCMS Data Differences - David WeilKhoranaNo ratings yet
- Eukaryotic Translation Initiation Factor 4EDocument8 pagesEukaryotic Translation Initiation Factor 4ENeeraj KumarNo ratings yet
- 1 s2.0 S037907381100541X MainDocument9 pages1 s2.0 S037907381100541X MainIlenia BracagliaNo ratings yet
- Research ArticleDocument9 pagesResearch ArticleMinh Đức HoàngNo ratings yet
- Bioinformatics Exercises PrintDocument6 pagesBioinformatics Exercises Printalem010No ratings yet
- Package Phvid': R Topics DocumentedDocument14 pagesPackage Phvid': R Topics Documentedoana.denisas10No ratings yet
- Enzyme Purification (Chapter 1) : Why Purify Proteins ??Document15 pagesEnzyme Purification (Chapter 1) : Why Purify Proteins ??Mohanad JawadNo ratings yet
- Gcms PosterDocument1 pageGcms Postervijay2109No ratings yet
- Protein Purification - CACDocument21 pagesProtein Purification - CACAhammed Abu Dil100% (1)
- 11.bioinformatics Analysis of ProteinsDocument49 pages11.bioinformatics Analysis of ProteinsAlexis JenatzyNo ratings yet
- Structure The Spectrin-Actin Binding Site Erythrocyte Protein 4.1Document5 pagesStructure The Spectrin-Actin Binding Site Erythrocyte Protein 4.1Rabiatul AdawiyahNo ratings yet
- Tutorial On Microarray Analysis Using Bioconductor and R (Sample Study)Document2 pagesTutorial On Microarray Analysis Using Bioconductor and R (Sample Study)Coțovanu IulianNo ratings yet
- BIO 20-1 Third Long Exam Bioinformatics Engineering: - We Can Get The Amino Acid Sequence Using Single Letter CodeDocument4 pagesBIO 20-1 Third Long Exam Bioinformatics Engineering: - We Can Get The Amino Acid Sequence Using Single Letter CodeArjohn GeocalloNo ratings yet
- GcmsDocument3 pagesGcmsRista Susanti100% (1)
- Structure Based Drug Designing For Mycoplasmal PneumoniaDocument23 pagesStructure Based Drug Designing For Mycoplasmal PneumoniaBhavya Kumar SinghNo ratings yet
- Peptide Mapping Using: MaldiDocument17 pagesPeptide Mapping Using: MaldiKumaran ShanmugamNo ratings yet
- Decentralized Info-Mediaries Proprietary Limited Academic DocumentDocument3 pagesDecentralized Info-Mediaries Proprietary Limited Academic DocumentYesterdaysJamNo ratings yet
- 717 FullDocument3 pages717 FullVreeq VreechaNo ratings yet
- Mega Script T7 KitDocument33 pagesMega Script T7 KitkitamandaNo ratings yet
- MudPit - ArtigoDocument4 pagesMudPit - ArtigoJoyce ReisNo ratings yet
- Lab No 4 - Affinity ChromatographyDocument8 pagesLab No 4 - Affinity Chromatographydead_knightNo ratings yet
- Practical Quantitative Biomedical Applications of MALDI-TOF Mass SpectrometryDocument13 pagesPractical Quantitative Biomedical Applications of MALDI-TOF Mass SpectrometryDiana ReyNo ratings yet
- Module 2 Overview: Spring BreakDocument16 pagesModule 2 Overview: Spring BreakHaripriya SantoshNo ratings yet
- Anakon 2001Document1 pageAnakon 2001AliAliNo ratings yet
- Index: Index Terms Links ADocument11 pagesIndex: Index Terms Links AAliAliNo ratings yet
- Immunoassay For Estrogens in The Environment: Multi-Analyte - Detektion Based On FluorescenceDocument1 pageImmunoassay For Estrogens in The Environment: Multi-Analyte - Detektion Based On FluorescenceAliAliNo ratings yet
- Front Matter1Document1 pageFront Matter1AliAliNo ratings yet
- Preface About The Author: Wastes BODDocument6 pagesPreface About The Author: Wastes BODAliAliNo ratings yet
- Design Calc - Cooling Tower Sizing - R1 - 15.10.2011Document1 pageDesign Calc - Cooling Tower Sizing - R1 - 15.10.2011AliAliNo ratings yet
- FPSBa 8cDocument1 pageFPSBa 8cAliAliNo ratings yet
- Water Treatment - Overview Ion ExchangeDocument9 pagesWater Treatment - Overview Ion ExchangeAliAliNo ratings yet
- In Our 2009-2010 Project, We Have Successfully Developed A Novel Method Which Combines ImmunomagneticDocument2 pagesIn Our 2009-2010 Project, We Have Successfully Developed A Novel Method Which Combines ImmunomagneticAliAliNo ratings yet
- Material Stream: F-G: ConditionsDocument2 pagesMaterial Stream: F-G: ConditionsAliAliNo ratings yet
- Fpsne 100Document1 pageFpsne 100AliAliNo ratings yet
- FPSMS 90Document1 pageFPSMS 90AliAliNo ratings yet
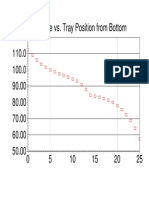
- 120.0 Temperature vs. Tray Position From BottomDocument1 page120.0 Temperature vs. Tray Position From BottomAliAliNo ratings yet
- Fps Magna Cide 575Document1 pageFps Magna Cide 575AliAliNo ratings yet
- FPSXL 2LDocument1 pageFPSXL 2LAliAliNo ratings yet
- Important Compatability Superlasticizes Portland CementDocument23 pagesImportant Compatability Superlasticizes Portland CementAliAliNo ratings yet
- 6520-3902004РТ ENGL. (ЕВРО -2, 3, 4)Document1,445 pages6520-3902004РТ ENGL. (ЕВРО -2, 3, 4)kiên HoangNo ratings yet
- CPE-214 Computer-Aided Engineering Design - Lab - Manual - OBE - 2 PDFDocument64 pagesCPE-214 Computer-Aided Engineering Design - Lab - Manual - OBE - 2 PDFHamza RaufNo ratings yet
- Seal Kit Seal KitDocument61 pagesSeal Kit Seal KitМаксим Стратила0% (1)
- Norton Introduction To Philosophy 2nd Edition Rosen Test BankDocument9 pagesNorton Introduction To Philosophy 2nd Edition Rosen Test Bankbenjaminhernandezfecmxjzrdt100% (14)
- Asynchronous ActivityDocument1 pageAsynchronous ActivityJoanna Mae MendozaNo ratings yet
- Phillip Hudson: (813) - 334-3337 PSH5017@psu - EduDocument1 pagePhillip Hudson: (813) - 334-3337 PSH5017@psu - Edupsh5017No ratings yet
- GP125 - 200s Maintenance Manual-A520180913Document170 pagesGP125 - 200s Maintenance Manual-A520180913paulatclayxNo ratings yet
- LP40W LED Driver 2017Document4 pagesLP40W LED Driver 2017Daniel MéndezNo ratings yet
- Industrial Automation and Control Systems - Chapter 1 - Basic Concepts of MeasurementsDocument21 pagesIndustrial Automation and Control Systems - Chapter 1 - Basic Concepts of MeasurementsHasan IsmailNo ratings yet
- Survey of Mechanical Working: A. IntroductionDocument4 pagesSurvey of Mechanical Working: A. IntroductionAsif AhmedNo ratings yet
- 2 ID FansDocument43 pages2 ID Fansshubham vermaNo ratings yet
- Bitacora 5th Week #6Document1 pageBitacora 5th Week #6Onelbi RamosNo ratings yet
- Kinematics of A Novel Nine Degree of Freedom Configurable Gough-Stewart PlatformDocument19 pagesKinematics of A Novel Nine Degree of Freedom Configurable Gough-Stewart PlatformNeider NadidNo ratings yet
- Bia Report 13-97Document32 pagesBia Report 13-97JohnNo ratings yet
- 10D Unit-Coral Reefs PDFDocument14 pages10D Unit-Coral Reefs PDFIWAN KUNCORONo ratings yet
- CNC Programming Intro & Code PDFDocument127 pagesCNC Programming Intro & Code PDFAswath SridharNo ratings yet
- Computer Application in BusinessDocument3 pagesComputer Application in BusinessChegg UserNo ratings yet
- SPPU Report FormatDocument50 pagesSPPU Report FormatAmit Devidas AherNo ratings yet
- Void of The Course of The MoonDocument16 pagesVoid of The Course of The Moonsonaliforex1No ratings yet
- Find The Belt Length at A 72 in Distance Connected in Open Belt. The Pulley Diameters Are 6 and 12 inDocument4 pagesFind The Belt Length at A 72 in Distance Connected in Open Belt. The Pulley Diameters Are 6 and 12 inGeoffrey GolbequeNo ratings yet
- 13 DR Ajeet SinghDocument6 pages13 DR Ajeet SinghSuraj SutharNo ratings yet
- Tugas Mek I-9 OktDocument2 pagesTugas Mek I-9 OktAlifani SofiNo ratings yet
- Planer MachineDocument37 pagesPlaner Machinemechfame89% (9)
- HOUSEKEEPING - WEEK 1 UpdatedDocument5 pagesHOUSEKEEPING - WEEK 1 UpdatedMaria Rizza IlaganNo ratings yet
- Learning Shell Scripting With ZSH Sample ChapterDocument24 pagesLearning Shell Scripting With ZSH Sample ChapterPackt PublishingNo ratings yet
- To Parts CatalogDocument18 pagesTo Parts CatalogaliNo ratings yet
- Quiz 1 Distribusi Plus JawabanDocument6 pagesQuiz 1 Distribusi Plus JawabandimasfupNo ratings yet
- PhotoshopCC KBSCDocument4 pagesPhotoshopCC KBSCHusk EisbornNo ratings yet
- University of Wah Wah Engineering College Assignment # 1: (Reg No.Document2 pagesUniversity of Wah Wah Engineering College Assignment # 1: (Reg No.Hiba AliNo ratings yet
- Cambridge O Level: PHYSICS 5054/42Document16 pagesCambridge O Level: PHYSICS 5054/42Lapu LapuNo ratings yet