You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Academic Press Encyclopedia of Physical Science and Technology 3rd PolymerDocument339 pagesAcademic Press Encyclopedia of Physical Science and Technology 3rd PolymerBLUECOMANCHE100% (1)
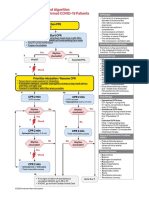
- AlgorithmPALS CACOVID 200406 PDFDocument1 pageAlgorithmPALS CACOVID 200406 PDFEka RahmanizarNo ratings yet
- ImmunologieDocument15 pagesImmunologieHaixing WangNo ratings yet
- Rosai DiseaseDocument9 pagesRosai DiseaseIrina Gabriela VizureanuNo ratings yet
- Immunology Exam Q's With AnswersDocument26 pagesImmunology Exam Q's With AnswersHavardKrovel87% (54)
- Immunology Core NotesDocument230 pagesImmunology Core NotesSabinaHorvatNo ratings yet
- Allergy ImmunotherapyDocument12 pagesAllergy Immunotherapyandreea g.No ratings yet
- Weight-For-Length GIRLS: Birth To 2 Years (Z-Scores)Document1 pageWeight-For-Length GIRLS: Birth To 2 Years (Z-Scores)Malisa LukmanNo ratings yet
- Maintain CVP ≥8 mm Hg in Septic ShockDocument6 pagesMaintain CVP ≥8 mm Hg in Septic ShockIbrahim DharmawanNo ratings yet
- Abstrak Anu Lampung Revisi FinalDocument1 pageAbstrak Anu Lampung Revisi FinalNisa UcilNo ratings yet
- HSR - USCOM FinalDocument49 pagesHSR - USCOM FinalNisa UcilNo ratings yet
- Perinatology Journal Reading by AhimsaDocument31 pagesPerinatology Journal Reading by AhimsaNisa UcilNo ratings yet
- Airway Management Complications in Children With Difficult Tracheal Intubation From The Pediatric Difficult Intubation PeDI Registry A Prospective Cohort AnalysisDocument10 pagesAirway Management Complications in Children With Difficult Tracheal Intubation From The Pediatric Difficult Intubation PeDI Registry A Prospective Cohort AnalysisNisa UcilNo ratings yet
- Airway Management Complications in Children With Difficult Tracheal Intubation From The Pediatric Difficult Intubation PeDI Registry A Prospective Cohort AnalysisDocument10 pagesAirway Management Complications in Children With Difficult Tracheal Intubation From The Pediatric Difficult Intubation PeDI Registry A Prospective Cohort AnalysisNisa UcilNo ratings yet
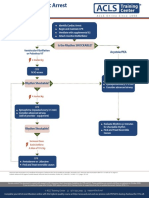
- Algo Pals BLS Pediatric Cardiac ArrestDocument1 pageAlgo Pals BLS Pediatric Cardiac ArrestSiti NabilaNo ratings yet
- Exercise During Pregnancy A Practical ApproachDocument7 pagesExercise During Pregnancy A Practical ApproachNisa UcilNo ratings yet
- NeurologicDocument7 pagesNeurologicFarrah ErmanNo ratings yet
- Carrot, Angel and Oreo cake recipesDocument4 pagesCarrot, Angel and Oreo cake recipesNisa UcilNo ratings yet
- Algo Pals Pediatric BradycardiaDocument1 pageAlgo Pals Pediatric BradycardiaNisa UcilNo ratings yet
- Infantile Pityriasis Alba and Comorbid DisordersDocument5 pagesInfantile Pityriasis Alba and Comorbid DisordersNisa UcilNo ratings yet
- Nihms 701642Document10 pagesNihms 701642Nisa UcilNo ratings yet
- The Prevalence of Interatrial Septal Openings in Newborns and Predictive Factors For Spontaneous ClosureDocument5 pagesThe Prevalence of Interatrial Septal Openings in Newborns and Predictive Factors For Spontaneous ClosureNisa UcilNo ratings yet
- Algo Pals Pediatric Cardiac ArrestDocument1 pageAlgo Pals Pediatric Cardiac ArrestDevi ChrestellaNo ratings yet
- Review Article: Autoimmune/Inflammatory Arthritis Associated Lymphomas: Who Is at Risk?Document12 pagesReview Article: Autoimmune/Inflammatory Arthritis Associated Lymphomas: Who Is at Risk?Nisa UcilNo ratings yet
- Exercise During Pregnancy A Practical ApproachDocument7 pagesExercise During Pregnancy A Practical ApproachNisa UcilNo ratings yet
- Early detection of autism with CHAT checklistDocument44 pagesEarly detection of autism with CHAT checklistNisa UcilNo ratings yet
- Adverson, J. 2013. Feeding Children With CP Swallowing Difficulties PDFDocument4 pagesAdverson, J. 2013. Feeding Children With CP Swallowing Difficulties PDFNicoleOrtegaAguileraNo ratings yet
- Maternal and Infant Risk Factors Associated with Neonatal Asphyxia in BaliDocument6 pagesMaternal and Infant Risk Factors Associated with Neonatal Asphyxia in BaliNisa UcilNo ratings yet
- Isk AafpDocument7 pagesIsk Aafpbebekdd22No ratings yet
- Cerebrospinal Fluid Lactate and PyruvateDocument8 pagesCerebrospinal Fluid Lactate and PyruvateNisa UcilNo ratings yet
- 66b3 PDFDocument6 pages66b3 PDFNisa UcilNo ratings yet
- Karagol 2010Document4 pagesKaragol 2010Nisa UcilNo ratings yet
- Isk AafpDocument7 pagesIsk Aafpbebekdd22No ratings yet
- Correlation of Procalcitonin Level and Neutrophil-Lymphocyte Ratio in SIRS PatientsDocument1 pageCorrelation of Procalcitonin Level and Neutrophil-Lymphocyte Ratio in SIRS PatientsNisa UcilNo ratings yet
- PCH 32 s1 039 PDFDocument4 pagesPCH 32 s1 039 PDF79lalalaNo ratings yet
- Validity and ReliabilityDocument6 pagesValidity and ReliabilityfarlynzNo ratings yet
- Microbiology exam questions and answersDocument58 pagesMicrobiology exam questions and answersAbhani MøhitNo ratings yet
- Pre-Medical Division: Immunity and DiseaseDocument55 pagesPre-Medical Division: Immunity and DiseaseAravind RaoNo ratings yet
- Effect of High Dose Vitamin C On Epstein-Barr Viral InfectionDocument8 pagesEffect of High Dose Vitamin C On Epstein-Barr Viral InfectionFilipos ConstantinNo ratings yet
- 2.2 Cell ContructionDocument25 pages2.2 Cell ContructionDesyNo ratings yet
- Mmunoglobulin: Sagar Sharma B.SC - MLT 4 SemDocument20 pagesMmunoglobulin: Sagar Sharma B.SC - MLT 4 SemSagar Sharma100% (1)
- Report for Vuyyooru Harish Kumar ReddyDocument5 pagesReport for Vuyyooru Harish Kumar ReddyKiran ReddyNo ratings yet
- Monoclonal AntibodiesDocument35 pagesMonoclonal Antibodiessam143143143143No ratings yet
- Laboratory Results RosarioDocument6 pagesLaboratory Results RosarioAlexander Eli BeytonNo ratings yet
- Homeopathy and ImmunologyDocument65 pagesHomeopathy and Immunologysunsign100% (2)
- Considerations For The Design of Antibody-Based TherapeuticsDocument30 pagesConsiderations For The Design of Antibody-Based TherapeuticsDaniel CastanNo ratings yet
- Chorus Kit List Rev.03 enDocument6 pagesChorus Kit List Rev.03 enHaitham Salama GhareebNo ratings yet
- Measles Virus Infection Diminishes Preexisting Antibodies That Offer Protection From Other Pathogens 599.fullDocument9 pagesMeasles Virus Infection Diminishes Preexisting Antibodies That Offer Protection From Other Pathogens 599.fullEngkiong GoNo ratings yet
- Idsa Covid 19 GL Serology v1.0Document66 pagesIdsa Covid 19 GL Serology v1.0Max CamargoNo ratings yet
- Felix Widal TestDocument9 pagesFelix Widal TestGaluhAlvianaNo ratings yet
- Antigen & Antibody: Structure, Function & Immune ResponseDocument48 pagesAntigen & Antibody: Structure, Function & Immune Responsebook keedaNo ratings yet
- Immunology: Dr. A.K.M. Akbar KabirDocument23 pagesImmunology: Dr. A.K.M. Akbar KabirRakib's exploration worldNo ratings yet
- Immunodiagnosis of Tuberculosis: An Update on Antibody Detection MethodsDocument5 pagesImmunodiagnosis of Tuberculosis: An Update on Antibody Detection MethodsPROFE EFRA LAGRANGENo ratings yet
- Immunoglobulins: Prof - Dr.Gülden Burçak 2020-2021Document25 pagesImmunoglobulins: Prof - Dr.Gülden Burçak 2020-2021Marwa AliNo ratings yet
- Extended Criteria For Organ Acceptance Strategies ClinTransplantDocument17 pagesExtended Criteria For Organ Acceptance Strategies ClinTransplantRicardo Ortiz NovilloNo ratings yet
- The Etiology of Glomerulonephritis: Roles of Infection and AutoimmunityDocument10 pagesThe Etiology of Glomerulonephritis: Roles of Infection and AutoimmunityCarlos DominguezNo ratings yet
- Joseph Irudhiyaraj Biomedical NanosensorDocument188 pagesJoseph Irudhiyaraj Biomedical NanosensorchandranNo ratings yet
- McqsDocument10 pagesMcqsDAWOODNo ratings yet
- BP605T Unit 1-3Document127 pagesBP605T Unit 1-3Gyampoh SolomonNo ratings yet
- Gen Bio 2 Q4 Module 4Document25 pagesGen Bio 2 Q4 Module 4Shoto TodorokiNo ratings yet