You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- E Book Secrets of Third Eye AwakeningDocument50 pagesE Book Secrets of Third Eye AwakeningKonkman100% (4)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5795)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Heat Transfer Through Pipe Support PDFDocument21 pagesHeat Transfer Through Pipe Support PDFSteve IpNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Creep ABAQUS PDFDocument68 pagesCreep ABAQUS PDFAdisa Tutusic100% (1)
- Advanced Design of Steel StructuresDocument3 pagesAdvanced Design of Steel StructuresManish Shashikant DharekNo ratings yet
- DSMTS-0080.6 Martensitic SS PowderDocument3 pagesDSMTS-0080.6 Martensitic SS PowderdespinozatNo ratings yet
- Construction Stage Analysis Reflecting Long-Term DeformationDocument18 pagesConstruction Stage Analysis Reflecting Long-Term DeformationAshish LoyaNo ratings yet
- TJ Lin RheologyDocument39 pagesTJ Lin RheologyKonkmanNo ratings yet
- Why SufferingDocument50 pagesWhy SufferingKonkman100% (1)
- End Disc and Shell ThicknessDocument16 pagesEnd Disc and Shell Thicknesssigit100% (1)
- Solution Manual For Chemical Engineering ThermodynamicsDocument4 pagesSolution Manual For Chemical Engineering ThermodynamicsBayu Prayudi Wibowo8% (36)
- Multi-Block Copolymers and Living PolymerisationDocument2 pagesMulti-Block Copolymers and Living PolymerisationAde AndriansyahNo ratings yet
- What? Me Worry!?! What? Me Worry!?!Document7 pagesWhat? Me Worry!?! What? Me Worry!?!KonkmanNo ratings yet
- En ASME ComparisonDocument18 pagesEn ASME ComparisonPeterWay100% (1)
- E Book Book of SurrenderDocument44 pagesE Book Book of SurrenderKonkmanNo ratings yet
- Introduction To Fluid Mechanics - James A. FayDocument66 pagesIntroduction To Fluid Mechanics - James A. FayPhanindra Attada100% (1)
- Bates, JPS, PH Dependent Disso Rate of Nitrofurantoin From Commercial Suspensions, Tablets and CapsulesDocument3 pagesBates, JPS, PH Dependent Disso Rate of Nitrofurantoin From Commercial Suspensions, Tablets and CapsulesKonkmanNo ratings yet
- GAD 3 2015 UncontrollabilityDocument10 pagesGAD 3 2015 UncontrollabilityKonkmanNo ratings yet
- Kelvin Equation - A ReviewDocument12 pagesKelvin Equation - A ReviewKonkmanNo ratings yet
- Coan & Shoichet, 2008 Stoichiometry and Physical Chemistry of Promiscuous AggregatorsDocument7 pagesCoan & Shoichet, 2008 Stoichiometry and Physical Chemistry of Promiscuous AggregatorsKonkmanNo ratings yet
- What? Me Worry!?! What? Me Worry!?!Document12 pagesWhat? Me Worry!?! What? Me Worry!?!KonkmanNo ratings yet
- E Book PYS 108 Truth of Yoga of EnlightenmentDocument277 pagesE Book PYS 108 Truth of Yoga of EnlightenmentKonkman100% (1)
- BS 2561carbon Fibre Reinforced Plastics UnidirecionalDocument16 pagesBS 2561carbon Fibre Reinforced Plastics UnidirecionalDamian GilNo ratings yet
- HW 3Document7 pagesHW 3Shashwat Chakraborti0% (1)
- Notes On Detailing Lab - CE692Document3 pagesNotes On Detailing Lab - CE692Barijit 65No ratings yet
- Exercise 8: Exact Solutions To Energy Equation Example 1: Energy Dissipation in Poiseuille FlowDocument4 pagesExercise 8: Exact Solutions To Energy Equation Example 1: Energy Dissipation in Poiseuille FlowRafael DalmauNo ratings yet
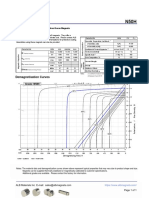
- N50H Grade Neodymium Magnets DataDocument1 pageN50H Grade Neodymium Magnets DataSteve HsuNo ratings yet
- Transport Phenomena Quiz 1 SolutionDocument2 pagesTransport Phenomena Quiz 1 SolutionAyman Al Kafrawy100% (1)
- Essentials of Materials Science and Engineering 3rd Edition Askeland Solutions Manual DownloadDocument9 pagesEssentials of Materials Science and Engineering 3rd Edition Askeland Solutions Manual DownloadDiana Grappe100% (26)
- Cse T Scds Kolonne enDocument16 pagesCse T Scds Kolonne enDeborah S. FructuosoNo ratings yet
- Finite Element Analysis of Base Isolated Buildings Subjected To Earthquake LoadsDocument21 pagesFinite Element Analysis of Base Isolated Buildings Subjected To Earthquake LoadsasdNo ratings yet
- Applsci 08 01960 PDFDocument17 pagesApplsci 08 01960 PDFphillynovitaNo ratings yet
- General Practice in Failure Analysis PDFDocument12 pagesGeneral Practice in Failure Analysis PDFAhmed AymanNo ratings yet
- Striclty No Erasures Allowed.: TEST II. MULTIPLE CHOICE: Encircle The Letter of The Correct AnswerDocument2 pagesStriclty No Erasures Allowed.: TEST II. MULTIPLE CHOICE: Encircle The Letter of The Correct AnswerKim Shai TanoNo ratings yet
- Polymers 14 03693 v2Document20 pagesPolymers 14 03693 v2Abd BAGHADNo ratings yet
- 墊子構裝Document17 pages墊子構裝Michael KaoNo ratings yet
- TA-Rayner-Canham6e ArtPPT Chapter08Document22 pagesTA-Rayner-Canham6e ArtPPT Chapter08Rey DLRNo ratings yet
- Fibre Reinforced Polymers - Strengths, Weaknesse, Opportunities and ThreatsDocument6 pagesFibre Reinforced Polymers - Strengths, Weaknesse, Opportunities and ThreatsalexisdiakNo ratings yet
- 19FTM14. 4D High Pressure Gas Quenching - A Leap in Performance vs. Press QuenchingDocument1 page19FTM14. 4D High Pressure Gas Quenching - A Leap in Performance vs. Press QuenchinggioNo ratings yet
- Ductile Iron: 2002 Issue 2Document47 pagesDuctile Iron: 2002 Issue 2karthikkandaNo ratings yet
- Phase 2 Test 4Document4 pagesPhase 2 Test 4Marcus PoonNo ratings yet
- Mechnotes: Unit - 1 ObjectiveDocument25 pagesMechnotes: Unit - 1 ObjectiveKaran SelvaNo ratings yet