You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Solid-Phase Microextraction of Volatile Components From Natural Grassland PlantsDocument7 pagesSolid-Phase Microextraction of Volatile Components From Natural Grassland PlantsZhang PinzhengNo ratings yet
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Data VisualisationDocument2 pagesData VisualisationZhang PinzhengNo ratings yet
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Revision FileDocument16 pagesRevision FileZhang PinzhengNo ratings yet
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
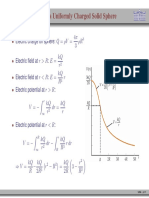
- Term Test: PC1432 - Physics IIEDocument9 pagesTerm Test: PC1432 - Physics IIEZhang PinzhengNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- CM1501 Finals Section B AnswersDocument9 pagesCM1501 Finals Section B AnswersZhang PinzhengNo ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Gaskell HalDocument123 pagesGaskell Halarcher1638100% (2)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- C10K Organic Lecture 4Document28 pagesC10K Organic Lecture 4Zhang PinzhengNo ratings yet
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Synthesis of iron(III) EDTA complexDocument2 pagesSynthesis of iron(III) EDTA complexAbhijit FarakteNo ratings yet
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Formula-Sheet For Mid-Term TestDocument3 pagesFormula-Sheet For Mid-Term TestZhang PinzhengNo ratings yet
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The uniqueness of electric potential around conductorsDocument8 pagesThe uniqueness of electric potential around conductorsHarish S KiranNo ratings yet
- tsl94 PDFDocument1 pagetsl94 PDFZhang PinzhengNo ratings yet
- J 100880 A 020Document7 pagesJ 100880 A 020Zhang PinzhengNo ratings yet
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Case Study - Sustainable and Smart CitiesDocument58 pagesCase Study - Sustainable and Smart CitiesZhang PinzhengNo ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Callister Materials Science and Engineering - An Introduction 7e Solutions ManualDocument1,112 pagesCallister Materials Science and Engineering - An Introduction 7e Solutions ManualHemavathy Rt76% (33)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Defining Moment in BMTDocument2 pagesDefining Moment in BMTZhang PinzhengNo ratings yet
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- IRAS Temp Employment Application Form (JC)Document1 pageIRAS Temp Employment Application Form (JC)Zhang PinzhengNo ratings yet
- Statistics, 4th Edition by David Freedman, Robert PisaniDocument715 pagesStatistics, 4th Edition by David Freedman, Robert PisaniZhang Pinzheng89% (66)
- Whirlpools For Science DayDocument5 pagesWhirlpools For Science DayZhang PinzhengNo ratings yet
- Whirlpools For Science DayDocument5 pagesWhirlpools For Science DayZhang PinzhengNo ratings yet
- 7-8 Essay Skills Compre AnswersDocument5 pages7-8 Essay Skills Compre AnswersZhang PinzhengNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- UntitledDocument1 pageUntitledZhang PinzhengNo ratings yet
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- FDG Production-Theory Operations-FDG Synthesis Chemistr1Document25 pagesFDG Production-Theory Operations-FDG Synthesis Chemistr1Cesar Adolfo SanchezNo ratings yet
- Oversized Particles in Emulsified Asphalts (Sieve Test) : Standard Test Method ForDocument2 pagesOversized Particles in Emulsified Asphalts (Sieve Test) : Standard Test Method Forمحمد سليمان بن عمرNo ratings yet
- Signature RedactedDocument49 pagesSignature RedactedG Pavan KumarNo ratings yet
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- HW1Document8 pagesHW1Anonymous fXSlye100% (1)
- Hough Transform in Matlab: - If We Find An Edge Point at (Ix, Iy), We Loop Through All Possible Values of ThetaDocument11 pagesHough Transform in Matlab: - If We Find An Edge Point at (Ix, Iy), We Loop Through All Possible Values of ThetaLe QuyenNo ratings yet
- DPP-1 QuantizationDocument1 pageDPP-1 QuantizationVikasNo ratings yet
- Midas FEADocument2 pagesMidas FEACristian Camilo Londoño PiedrahítaNo ratings yet
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- P 211enDocument26 pagesP 211enRadhakrishnan BalasubramanianNo ratings yet
- A Study of Manufacturing of Steam TurbinesDocument40 pagesA Study of Manufacturing of Steam TurbinesSaketh Varma MudunuriNo ratings yet
- Q3 ST 1 GR.6 Science With TosDocument4 pagesQ3 ST 1 GR.6 Science With TosRed MariposaNo ratings yet
- Introducing JiFi ST Petersburg 2014Document4 pagesIntroducing JiFi ST Petersburg 2014danjohhnNo ratings yet
- Pivot Interactives Motion Graphing A Dry Ice Puck On A RampDocument2 pagesPivot Interactives Motion Graphing A Dry Ice Puck On A RampSophia0% (1)
- EPA 1668 A, Ag-2003Document129 pagesEPA 1668 A, Ag-2003Karina Rondon RivadeneyraNo ratings yet
- Terjemahan BukuDocument2 pagesTerjemahan BukuSeprianNo ratings yet
- Acids and Bases: Answers To Worked ExamplesDocument12 pagesAcids and Bases: Answers To Worked ExamplesDana CapbunNo ratings yet
- Adrian Stan MFQMCourseHsL2006Document60 pagesAdrian Stan MFQMCourseHsL2006禿公No ratings yet
- cO2CH4 Permselective GassensorDocument5 pagescO2CH4 Permselective GassensorKartik RamasubramanianNo ratings yet
- Schrodinger Equation DerivationDocument12 pagesSchrodinger Equation DerivationAndrés López Martínez100% (1)
- S P I C e J e T Q U e S T I o N SDocument43 pagesS P I C e J e T Q U e S T I o N SDharavGosaliaNo ratings yet
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- General Physics 1 1st Quarter Module 1 ActivitiesDocument16 pagesGeneral Physics 1 1st Quarter Module 1 ActivitiesMica LopezNo ratings yet
- Design and Manufacturing of Automatic Gear Shifter For BicycleDocument10 pagesDesign and Manufacturing of Automatic Gear Shifter For BicycleMannam RujendraNo ratings yet
- Phased Array Probes and Wedges: Probe CatalogDocument3 pagesPhased Array Probes and Wedges: Probe CatalogDavidMontillaNo ratings yet
- Welding Journal PDFDocument7 pagesWelding Journal PDFraisalfiansyahNo ratings yet
- Manifest Your Desires with The Quantum CookbookDocument14 pagesManifest Your Desires with The Quantum CookbookAgarta1111No ratings yet
- Dosing Pump Innovata Drive ConceptDocument5 pagesDosing Pump Innovata Drive ConceptgarpNo ratings yet
- Reability PDFDocument396 pagesReability PDFMarcelo Ziulkoski100% (1)
- Ordinary Differential EquationDocument20 pagesOrdinary Differential EquationRadeanindaNo ratings yet
- Stiffness of Cable-Based Parallel Manipulators With Application To Stability AnalysisDocument8 pagesStiffness of Cable-Based Parallel Manipulators With Application To Stability AnalysisNhật MinhNo ratings yet
- Steam Engineering Principles and Heat TransferDocument99 pagesSteam Engineering Principles and Heat Transferalex mobileNo ratings yet
- Functional Safety from Scratch: A Practical Guide to Process Industry ApplicationsFrom EverandFunctional Safety from Scratch: A Practical Guide to Process Industry ApplicationsNo ratings yet
- Guidelines for Chemical Process Quantitative Risk AnalysisFrom EverandGuidelines for Chemical Process Quantitative Risk AnalysisRating: 5 out of 5 stars5/5 (1)