You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Mitral Stenosis Before, During and After PregnancyDocument6 pagesMitral Stenosis Before, During and After PregnancyAnna AlthafunnisaNo ratings yet
- Nephrotic Syndrome in AdultDocument53 pagesNephrotic Syndrome in AdultAnna Althafunnisa100% (1)
- General Cultural Scholarship Scheme 2015 - 2016: Check ListDocument7 pagesGeneral Cultural Scholarship Scheme 2015 - 2016: Check ListAnna AlthafunnisaNo ratings yet
- General Instructions To Applicants 2015-2016Document6 pagesGeneral Instructions To Applicants 2015-2016Anna AlthafunnisaNo ratings yet
- University List Under ICCR Scholarship Programme PDFDocument4 pagesUniversity List Under ICCR Scholarship Programme PDFAnna AlthafunnisaNo ratings yet
- Kunci Jawaban TOEFL Prediction Test 2Document2 pagesKunci Jawaban TOEFL Prediction Test 2Anna AlthafunnisaNo ratings yet
- SOEKARNODocument2 pagesSOEKARNOAnna AlthafunnisaNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Bright Adjei B.A. (Hons.)Document136 pagesBright Adjei B.A. (Hons.)gideon A. owusuNo ratings yet
- Newborn Screening of Neuromuscular Diseases - 2021 - Neuromuscular DisordersDocument11 pagesNewborn Screening of Neuromuscular Diseases - 2021 - Neuromuscular DisordersSuzie Simone Mardones SilvaNo ratings yet
- Asma Felino TratamientoDocument7 pagesAsma Felino TratamientoRenzo Alessandro Pacherres NietoNo ratings yet
- Pat An AnewDocument126 pagesPat An AnewgshchurovskiyNo ratings yet
- Drugs-1.ppt 0Document22 pagesDrugs-1.ppt 0Esraa BahaaNo ratings yet
- The Sovereign Remedy: Touch-Pieces and The King's Evil. (Pt. I) / Noel WoolfDocument30 pagesThe Sovereign Remedy: Touch-Pieces and The King's Evil. (Pt. I) / Noel WoolfDigital Library Numis (DLN)No ratings yet
- Food Safety Officer and Technical Officer FSSAI Guide 2019 PDFDocument15 pagesFood Safety Officer and Technical Officer FSSAI Guide 2019 PDFChinmoy Das0% (5)
- Meal Plan Medyo FinalDocument14 pagesMeal Plan Medyo FinalJasha MaeNo ratings yet
- Barneys Farm SeedsDocument46 pagesBarneys Farm SeedsPedro MarquesNo ratings yet
- Genitourinary SystemDocument27 pagesGenitourinary SystemDanica FrancoNo ratings yet
- Supreme Court: Republic of The Philippines Manila Third DivisionDocument7 pagesSupreme Court: Republic of The Philippines Manila Third DivisionRaymond RogacionNo ratings yet
- Tog 12228Document7 pagesTog 12228Avanie PalNo ratings yet
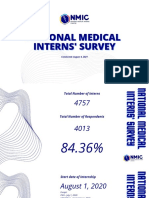
- National Medical National Medical Interns' Survey Interns' SurveyDocument20 pagesNational Medical National Medical Interns' Survey Interns' SurveyKaren Jodes CapananNo ratings yet
- Modalitas RehabmedikDocument115 pagesModalitas RehabmedikWegrimel AriegaraNo ratings yet
- Ahmed Proposal2Document34 pagesAhmed Proposal2ahmedabdikerimNo ratings yet
- Brochure ESSENZA en 05-2008Document20 pagesBrochure ESSENZA en 05-2008hgaucherNo ratings yet
- Sweet Potato Balls 1Document13 pagesSweet Potato Balls 1Rosha Camille100% (4)
- DSE-03-U2 - Behavior & HealthDocument23 pagesDSE-03-U2 - Behavior & HealthIndrashis MandalNo ratings yet
- Summary Notes - Topic 9 Transport in Animals - CIE Biology IGCSEDocument5 pagesSummary Notes - Topic 9 Transport in Animals - CIE Biology IGCSEMadhan Reddy SuddalaNo ratings yet
- Saudi License Exam (SLE) For General Dentistry - Sep 6, 2011Document10 pagesSaudi License Exam (SLE) For General Dentistry - Sep 6, 2011asifhameed90% (10)
- Pharmacology of Drugs Acting On Renal SystemDocument20 pagesPharmacology of Drugs Acting On Renal SystemBestha Chakri100% (2)
- Toshiba Xario XG User ManualDocument25 pagesToshiba Xario XG User ManualSalah AnamNo ratings yet
- Answers Part 2Document64 pagesAnswers Part 2Rynjeff Lui-Pio100% (1)
- US SchubertDocument21 pagesUS SchubertNirmayi HomkarNo ratings yet
- MainDocument10 pagesMainAnna SamsudinNo ratings yet
- School Letter Governor Carney 03132020Document2 pagesSchool Letter Governor Carney 03132020KevinOhlandtNo ratings yet
- Latihan Inggris SNMPTN 2012 Kode734Document4 pagesLatihan Inggris SNMPTN 2012 Kode734Pamela FoxNo ratings yet
- Drug Study GabapentinDocument3 pagesDrug Study Gabapentinbridget.badiang001No ratings yet
- Pharmacology, Pathology, Genetics: QP Code: BNN203Document1 pagePharmacology, Pathology, Genetics: QP Code: BNN203Mamta KumariNo ratings yet
- The Health Belief ModelDocument13 pagesThe Health Belief ModelSteph DayaganonNo ratings yet