You might also like
- Analytical Characterization of BiotherapeuticsFrom EverandAnalytical Characterization of BiotherapeuticsJennie R. LillNo ratings yet
- Epa 6010cDocument30 pagesEpa 6010cjuli_radNo ratings yet
- Power Transformer Online Monitoring Using Electromagnetic WavesFrom EverandPower Transformer Online Monitoring Using Electromagnetic WavesNo ratings yet
- EPA Method 6020 Heavy MetalsDocument18 pagesEPA Method 6020 Heavy MetalsMike CoronaNo ratings yet
- Power Electronics Applied to Industrial Systems and Transports, Volume 4: Electromagnetic CompatibilityFrom EverandPower Electronics Applied to Industrial Systems and Transports, Volume 4: Electromagnetic CompatibilityNo ratings yet
- Epa-Method-6020a Inductively Coupled Plasma-Mass SpectrometryDocument30 pagesEpa-Method-6020a Inductively Coupled Plasma-Mass SpectrometrydavidandrasiNo ratings yet
- Partial-Update Adaptive Signal Processing: Design Analysis and ImplementationFrom EverandPartial-Update Adaptive Signal Processing: Design Analysis and ImplementationNo ratings yet
- Astm d5673 1996Document9 pagesAstm d5673 1996luisry1990No ratings yet
- Trace Element Analysis of Water and WastesDocument18 pagesTrace Element Analysis of Water and WastesElprince MidoNo ratings yet
- E60 Analysis of Metals, Ores, and Related Materials by Molecular Absorption Spectrometry1Document8 pagesE60 Analysis of Metals, Ores, and Related Materials by Molecular Absorption Spectrometry1Electra AkbarillahNo ratings yet
- E275-08 (2013) Standard Practice For Describing and Measuring Performance of Ultraviolet and Visible SpectrophotometersDocument11 pagesE275-08 (2013) Standard Practice For Describing and Measuring Performance of Ultraviolet and Visible Spectrophotometersislamaktham100% (3)
- Determining Relative Spectral Correction Factors For Emission Signal of Fluorescence SpectrometersDocument5 pagesDetermining Relative Spectral Correction Factors For Emission Signal of Fluorescence Spectrometersmohammed karasnehNo ratings yet
- E572-13 - Standard Test Method For Analysis of Stainless and Alloy Steels by Wavelength Dispersive X-Ray Fluorescence Spectrometry.Document11 pagesE572-13 - Standard Test Method For Analysis of Stainless and Alloy Steels by Wavelength Dispersive X-Ray Fluorescence Spectrometry.Royce BergNo ratings yet
- Astm E0169 16Document6 pagesAstm E0169 16archanaNo ratings yet
- E 578 - 01 - Rtu3oa - PDFDocument3 pagesE 578 - 01 - Rtu3oa - PDFMichelle ZacariaNo ratings yet
- E 578 - 83 R98 - Rtu3oc1sruqDocument4 pagesE 578 - 83 R98 - Rtu3oc1sruqpechugonisNo ratings yet
- Epa 8323Document22 pagesEpa 8323Jane Ligia GramkowNo ratings yet
- Analysis of Metals, Ores, and Related Materials by SpectrophotometryDocument8 pagesAnalysis of Metals, Ores, and Related Materials by SpectrophotometryShehrije BejtaNo ratings yet
- Optical Emission Spectroscopy As An Analytical ToolDocument27 pagesOptical Emission Spectroscopy As An Analytical ToolJayee NonickNo ratings yet
- Osha Id 125gDocument43 pagesOsha Id 125gZenal AbidinNo ratings yet
- Osha Id 125gDocument43 pagesOsha Id 125gapriliandiniNo ratings yet
- C 1111 - 98 - QzexmtetotgDocument5 pagesC 1111 - 98 - Qzexmtetotggrats_singcoNo ratings yet
- 2 03 Counting Errors and Statistics SGDocument49 pages2 03 Counting Errors and Statistics SGPataki SandorNo ratings yet
- Oxygen Content Using A 14-Mev Neutron Activation and Direct-Counting TechniqueDocument8 pagesOxygen Content Using A 14-Mev Neutron Activation and Direct-Counting Techniqueruben carcamoNo ratings yet
- E578-07 (2013) Standard Test Method For Linearity of Fluorescence Measuring SystemsDocument3 pagesE578-07 (2013) Standard Test Method For Linearity of Fluorescence Measuring SystemsislamakthamNo ratings yet
- Astm E2534 20Document7 pagesAstm E2534 20Mohamed AboelkhierNo ratings yet
- D6443Document7 pagesD6443rimi7al100% (1)
- Standard Horizontal Icp-Aes Method Determination of Dissolved Elements by Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES)Document10 pagesStandard Horizontal Icp-Aes Method Determination of Dissolved Elements by Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES)Shahad A. YounisNo ratings yet
- F 358 - 83 R96 - Rjm1oc04m1i5nkuxDocument4 pagesF 358 - 83 R96 - Rjm1oc04m1i5nkuxjamaljamal20No ratings yet
- Metals by Plasma Emission SpectrosDocument8 pagesMetals by Plasma Emission Spectrosnhafa1311No ratings yet
- Analytical ChemistryDocument4 pagesAnalytical ChemistryAbdurrazaqNo ratings yet
- E 275 Â " 93 RTI3NS05MWDocument10 pagesE 275 Â " 93 RTI3NS05MWmario valenzuelaNo ratings yet
- Open-Path Fourier Transform Infrared (OP/FT-IR) Monitoring of Gases and Vapors in AirDocument19 pagesOpen-Path Fourier Transform Infrared (OP/FT-IR) Monitoring of Gases and Vapors in AirEric GozzerNo ratings yet
- UNIT: SpectrophotometryDocument15 pagesUNIT: SpectrophotometrybiddyusmcNo ratings yet
- New Optimized Analysis Method For Measuring Extended Grounding SystemsDocument4 pagesNew Optimized Analysis Method For Measuring Extended Grounding SystemsKiriakos SiderakisNo ratings yet
- E 275 Â " 93 RTI3NS1SRUQDocument12 pagesE 275 Â " 93 RTI3NS1SRUQmario valenzuelaNo ratings yet
- ASTM 2491 - 08 PhaseArray PDFDocument18 pagesASTM 2491 - 08 PhaseArray PDFOmero MeraNo ratings yet
- F 2405 - 04 Rji0mduDocument5 pagesF 2405 - 04 Rji0mdunooralhudNo ratings yet
- Uv Vis Spectroscopy ScattererDocument8 pagesUv Vis Spectroscopy ScatterercaespindoNo ratings yet
- Transmission Line Signature AnalysisDocument6 pagesTransmission Line Signature AnalysisLalit KumarNo ratings yet
- Composition SensorsDocument13 pagesComposition SensorsJorge Huerta100% (1)
- ASTM D 7085 - 04 (2010) E1Document10 pagesASTM D 7085 - 04 (2010) E1quochunguicNo ratings yet
- X-Ray Spectrometric Analysis of Lime and Limestone: Standard Test Method ForDocument5 pagesX-Ray Spectrometric Analysis of Lime and Limestone: Standard Test Method Forbaher74No ratings yet
- Method 300-1 1997Document40 pagesMethod 300-1 1997Sofy TaylorNo ratings yet
- General Techniques of Ultraviolet-Visible Quantitative AnalysisDocument6 pagesGeneral Techniques of Ultraviolet-Visible Quantitative Analysis陳明書No ratings yet
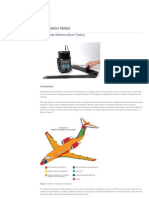
- Application Notes: Multi-Mode Adhesive Bond TestingDocument10 pagesApplication Notes: Multi-Mode Adhesive Bond TestingPDDELUCANo ratings yet
- Astm e 572 Rev A 2002 e 2006 R 2006Document10 pagesAstm e 572 Rev A 2002 e 2006 R 2006Jorge ToribioNo ratings yet
- Methods of Estimation of MultiDocument41 pagesMethods of Estimation of Multiapi-19786321100% (1)
- Describing and Specifying A Direct Current Plasma Atomic Emission SpectrometerDocument6 pagesDescribing and Specifying A Direct Current Plasma Atomic Emission SpectrometerEric GozzerNo ratings yet
- Electronic Measurements & Instrumentation 3 PDFDocument17 pagesElectronic Measurements & Instrumentation 3 PDFdenaiNo ratings yet
- Sakai Chapter 3Document20 pagesSakai Chapter 3Ronnel Mendoza VasquezNo ratings yet
- QC Lec Describe The Following Parts of The Apparatus For HPLC A) Column B) Pump C) Injection D) Detector E) ComputerDocument11 pagesQC Lec Describe The Following Parts of The Apparatus For HPLC A) Column B) Pump C) Injection D) Detector E) ComputerWillie HolcombNo ratings yet
- Practical Eddy Current Testing: - General ProcedureDocument7 pagesPractical Eddy Current Testing: - General Procedurekhizer mohamedNo ratings yet
- Thermo Beer LambertDocument2 pagesThermo Beer LambertRizka Rinda PramastiNo ratings yet
- Tin Oxide Gas Sensing: Comparison Among Different Measurement Techniques For Gas Mixture ClassificationDocument5 pagesTin Oxide Gas Sensing: Comparison Among Different Measurement Techniques For Gas Mixture ClassificationMohammad Kabir HossainNo ratings yet
- Impedance Spectroscopy and Experimental SetupDocument18 pagesImpedance Spectroscopy and Experimental SetupJako SibueaNo ratings yet
- E 169 - 04 (2014) PDFDocument6 pagesE 169 - 04 (2014) PDFruben carcamoNo ratings yet
- RTK1OADocument5 pagesRTK1OAwpwmhatNo ratings yet
- ASTM Guide 1588E-10Document5 pagesASTM Guide 1588E-10maraid sosaNo ratings yet
- Science of The Total EnvironmentDocument11 pagesScience of The Total EnvironmentSubs KatsNo ratings yet
- Detailed Complete List of NNRMS ProjectsDocument17 pagesDetailed Complete List of NNRMS ProjectsSubs KatsNo ratings yet
- Vegetable QuestionDocument47 pagesVegetable QuestionSubs KatsNo ratings yet
- Water ShedDocument10 pagesWater ShedSubs KatsNo ratings yet
- Viewpoint: Are Microbes at The Root of A Solution To World Food Production?Document5 pagesViewpoint: Are Microbes at The Root of A Solution To World Food Production?Subs KatsNo ratings yet
- Biomass and BiofuelsDocument19 pagesBiomass and BiofuelsSubs KatsNo ratings yet
- CXS 192eDocument396 pagesCXS 192eSubs KatsNo ratings yet
- For Office Use Only: Proposal NoDocument5 pagesFor Office Use Only: Proposal NoSubs KatsNo ratings yet
- Microbial Ecology: Opportunities For Inquiry-Based Learning: ASM CUE 2007 Try Something New WorkshopDocument21 pagesMicrobial Ecology: Opportunities For Inquiry-Based Learning: ASM CUE 2007 Try Something New WorkshopSubs KatsNo ratings yet
- Bioresource Technology: Yuyi Yang, Guan Wang, Bing Wang, Zeli Li, Xiaoming Jia, Qifa Zhou, Yuhua ZhaoDocument7 pagesBioresource Technology: Yuyi Yang, Guan Wang, Bing Wang, Zeli Li, Xiaoming Jia, Qifa Zhou, Yuhua ZhaoSubs KatsNo ratings yet
- Appl. Environ. Microbiol. 2012 Tymensen 4051 6Document7 pagesAppl. Environ. Microbiol. 2012 Tymensen 4051 6Subs KatsNo ratings yet
- Balance and Distribution of Sulphur in Volcanic Ash-Derived Soils in ChileDocument7 pagesBalance and Distribution of Sulphur in Volcanic Ash-Derived Soils in ChileSubs KatsNo ratings yet
- SolarGIS Imaps Sample ReportDocument2 pagesSolarGIS Imaps Sample ReportSubs KatsNo ratings yet
- Isolation of Cellulase Producing Fungi From Soil, Optimization and Molecular Characterization of The Isolate For Maximizing The Enzyme YieldDocument9 pagesIsolation of Cellulase Producing Fungi From Soil, Optimization and Molecular Characterization of The Isolate For Maximizing The Enzyme YieldSubs KatsNo ratings yet
- A Facultative Methanotroph For Environmental Remediation Applications A Facultative Methanotroph For Environmental Remediation ApplicationsDocument1 pageA Facultative Methanotroph For Environmental Remediation Applications A Facultative Methanotroph For Environmental Remediation ApplicationsSubs KatsNo ratings yet
- Instructions: CHE 314 - Heat Transfer Final Exam (Fall 2018), Dec. 18, 9:00am-11:00am: PavilionDocument8 pagesInstructions: CHE 314 - Heat Transfer Final Exam (Fall 2018), Dec. 18, 9:00am-11:00am: PavilionAkib ImtihanNo ratings yet
- Magnesium: Current and Alternative Production Routes: August 2010Document12 pagesMagnesium: Current and Alternative Production Routes: August 2010Salem GarrabNo ratings yet
- 5070 w17 Ms 42 PDFDocument9 pages5070 w17 Ms 42 PDFdR SHAMMIR AHMEDNo ratings yet
- Tetraamminecopper (II) Sulfate Monohydrate: Discover New Opportunities at Ultra High PurityDocument1 pageTetraamminecopper (II) Sulfate Monohydrate: Discover New Opportunities at Ultra High Purityjorge fravegaNo ratings yet
- Stainlesssteelforgings 9016Document46 pagesStainlesssteelforgings 9016Julian MësäNo ratings yet
- Batch Processing Solutions: For Oral Solid Dosage FormsDocument44 pagesBatch Processing Solutions: For Oral Solid Dosage FormsBulent Inan100% (1)
- SCION 456-GC: Specification SheetDocument8 pagesSCION 456-GC: Specification SheetNashiha SakinaNo ratings yet
- Guidelines For Ethylene Quench Tower Rev IntroDocument12 pagesGuidelines For Ethylene Quench Tower Rev IntroGuntoro AliNo ratings yet
- 5.1 Defects AllDocument40 pages5.1 Defects AllOmar Giovanny Ballén RodríguezNo ratings yet
- Chemical Aspect of Document ExaminationDocument6 pagesChemical Aspect of Document Examinationgenelord opalla100% (2)
- Cathodic Protection PresentationDocument56 pagesCathodic Protection Presentationsameer_rect2429100% (13)
- Heat Transfer in Olga 2000Document11 pagesHeat Transfer in Olga 2000Akin MuhammadNo ratings yet
- Toxicology Laboratory ManualDocument110 pagesToxicology Laboratory Manualnguyen ba trung100% (1)
- Me8391 Engineering ThermodynamicsDocument1 pageMe8391 Engineering ThermodynamicsAiam PandianNo ratings yet
- Reading2D Spectrum PDFDocument6 pagesReading2D Spectrum PDFRaihan Uchiha100% (1)
- David W L Hukins-X-ray diffraction by disordered and ordered systems _ covering X-ray diffraction by gases, liquids, and solids and indicating how the theory of diffraction by these different states .pdfDocument98 pagesDavid W L Hukins-X-ray diffraction by disordered and ordered systems _ covering X-ray diffraction by gases, liquids, and solids and indicating how the theory of diffraction by these different states .pdfJhan Carlos Bran Reyes100% (1)
- Basic Electronics1Document27 pagesBasic Electronics1api-291159108No ratings yet
- Discover The: PossibilitiesDocument16 pagesDiscover The: PossibilitiesFranz CruzNo ratings yet
- Problem Solving Laboratory (PSL)Document35 pagesProblem Solving Laboratory (PSL)Nurun Nadia MasromNo ratings yet
- Analysis Results Report E MDocument3 pagesAnalysis Results Report E MViorel PopNo ratings yet
- Course: Vikaas (Ja) - Batch:Ja: Resonance Eduventures LimitedDocument21 pagesCourse: Vikaas (Ja) - Batch:Ja: Resonance Eduventures LimiteddeepaktdeepakNo ratings yet
- Piccs 2012 PDFDocument1,614 pagesPiccs 2012 PDFAnnabelle GuilingNo ratings yet
- Materials System SpecificationDocument28 pagesMaterials System SpecificationAli RazaNo ratings yet
- Determination of Inoculum For Microbiological Testing: Micro Bio Lo Gy To Pic SDocument5 pagesDetermination of Inoculum For Microbiological Testing: Micro Bio Lo Gy To Pic SKenny JhodyNo ratings yet
- CF1FR v2.0 OnlineDocument2 pagesCF1FR v2.0 OnlineAnonymous 3VAQ9SNxl7No ratings yet
- Chemical Bonding SPECIAL ASSIGNMENTDocument25 pagesChemical Bonding SPECIAL ASSIGNMENTprexa indiaNo ratings yet
- Chapter 5 ComplexionDocument62 pagesChapter 5 ComplexionPHƯƠNG ĐẶNG YẾNNo ratings yet
- 50B-4KG1 Globe 2050B-4KG1 Angle: Fire Protection Pressure Relief ValveDocument2 pages50B-4KG1 Globe 2050B-4KG1 Angle: Fire Protection Pressure Relief ValveAlbeiro LeivaNo ratings yet
- Method StatementDocument5 pagesMethod StatementbecpavanNo ratings yet
- Q 0092r1 - Mastersizer 3000 MAZ6222 2022 Kimia FarmaDocument4 pagesQ 0092r1 - Mastersizer 3000 MAZ6222 2022 Kimia FarmaCapital ExpenditureNo ratings yet
- Power of Habit: The Ultimate Guide to Forming Positive Daily Habits, Learn How to Effectively Break Your Bad Habits For Good and Start Creating Good OnesFrom EverandPower of Habit: The Ultimate Guide to Forming Positive Daily Habits, Learn How to Effectively Break Your Bad Habits For Good and Start Creating Good OnesRating: 4.5 out of 5 stars4.5/5 (21)
- The Journeyman Electrician Exam Study Guide: Proven Methods for Successfully Passing the Journeyman Electrician Exam with ConfidenceFrom EverandThe Journeyman Electrician Exam Study Guide: Proven Methods for Successfully Passing the Journeyman Electrician Exam with ConfidenceNo ratings yet
- Well Integrity for Workovers and RecompletionsFrom EverandWell Integrity for Workovers and RecompletionsRating: 5 out of 5 stars5/5 (3)
- The Complete HVAC BIBLE for Beginners: The Most Practical & Updated Guide to Heating, Ventilation, and Air Conditioning Systems | Installation, Troubleshooting and Repair | Residential & CommercialFrom EverandThe Complete HVAC BIBLE for Beginners: The Most Practical & Updated Guide to Heating, Ventilation, and Air Conditioning Systems | Installation, Troubleshooting and Repair | Residential & CommercialNo ratings yet
- Building Energy Management Systems and Techniques: Principles, Methods, and ModellingFrom EverandBuilding Energy Management Systems and Techniques: Principles, Methods, and ModellingNo ratings yet
- Renewable Energy: Physics, Engineering, Environmental Impacts, Economics and PlanningFrom EverandRenewable Energy: Physics, Engineering, Environmental Impacts, Economics and PlanningRating: 5 out of 5 stars5/5 (4)
- Well Control for Completions and InterventionsFrom EverandWell Control for Completions and InterventionsRating: 4 out of 5 stars4/5 (10)
- Renewable Energy Finance: Theory and PracticeFrom EverandRenewable Energy Finance: Theory and PracticeRating: 4 out of 5 stars4/5 (1)
- Introduction to Power System ProtectionFrom EverandIntroduction to Power System ProtectionRating: 5 out of 5 stars5/5 (1)
- Electric Motor Control: DC, AC, and BLDC MotorsFrom EverandElectric Motor Control: DC, AC, and BLDC MotorsRating: 4.5 out of 5 stars4.5/5 (19)
- Oil: An Overview of the Petroleum IndustryFrom EverandOil: An Overview of the Petroleum IndustryRating: 4.5 out of 5 stars4.5/5 (3)
- The Homeowner's DIY Guide to Electrical WiringFrom EverandThe Homeowner's DIY Guide to Electrical WiringRating: 5 out of 5 stars5/5 (2)
- The Grid: The Fraying Wires Between Americans and Our Energy FutureFrom EverandThe Grid: The Fraying Wires Between Americans and Our Energy FutureRating: 3.5 out of 5 stars3.5/5 (48)
- Oil companies and the energy transitionFrom EverandOil companies and the energy transitionNo ratings yet
- Shorting the Grid: The Hidden Fragility of Our Electric GridFrom EverandShorting the Grid: The Hidden Fragility of Our Electric GridRating: 4.5 out of 5 stars4.5/5 (2)
- Air Cooled Heat Exchanger Handbook: Fundamentals, Calculations, Design and Q&AFrom EverandAir Cooled Heat Exchanger Handbook: Fundamentals, Calculations, Design and Q&ANo ratings yet
- Electric Motors and Drives: Fundamentals, Types and ApplicationsFrom EverandElectric Motors and Drives: Fundamentals, Types and ApplicationsRating: 4.5 out of 5 stars4.5/5 (12)
- Formulas and Calculations for Drilling, Production, and Workover: All the Formulas You Need to Solve Drilling and Production ProblemsFrom EverandFormulas and Calculations for Drilling, Production, and Workover: All the Formulas You Need to Solve Drilling and Production ProblemsNo ratings yet
- Handbook of Offshore Oil and Gas OperationsFrom EverandHandbook of Offshore Oil and Gas OperationsRating: 4.5 out of 5 stars4.5/5 (4)
- Case Studies of Material Corrosion Prevention for Oil and Gas ValvesFrom EverandCase Studies of Material Corrosion Prevention for Oil and Gas ValvesNo ratings yet
- Global Landscape of Renewable Energy FinanceFrom EverandGlobal Landscape of Renewable Energy FinanceNo ratings yet
- Natural Gas Processing: Technology and Engineering DesignFrom EverandNatural Gas Processing: Technology and Engineering DesignRating: 5 out of 5 stars5/5 (8)
- Thermal Power Plant: Design and OperationFrom EverandThermal Power Plant: Design and OperationRating: 4.5 out of 5 stars4.5/5 (23)
- Handbook on Battery Energy Storage SystemFrom EverandHandbook on Battery Energy Storage SystemRating: 4.5 out of 5 stars4.5/5 (2)