You might also like
- On The Relationship Between Field Cycling and Imprint in Ferroelectric Hf0.5Zr0.5O2Document9 pagesOn The Relationship Between Field Cycling and Imprint in Ferroelectric Hf0.5Zr0.5O2Hunsa WattanasasrnNo ratings yet
- Journal: Origin of The Potential Drop Over The Deposit During Electrophoretic DepositionDocument6 pagesJournal: Origin of The Potential Drop Over The Deposit During Electrophoretic DepositionMario Misael Machado LòpezNo ratings yet
- Besrapub 54Document8 pagesBesrapub 54Laxmidhar BesraNo ratings yet
- Adsorption: Solid ElectrodesDocument84 pagesAdsorption: Solid ElectrodesNemo NeroNo ratings yet
- Study of The Underlying Electrochemistry of Polycrystalline GoldDocument13 pagesStudy of The Underlying Electrochemistry of Polycrystalline GoldAzucena osornio villaNo ratings yet
- Hidrogeno - OxigenoDocument3 pagesHidrogeno - OxigenoVanessa BeltránNo ratings yet
- Advancement of Intermetallic Compounds and Combinations Generally Stayed and Their ReviewDocument2 pagesAdvancement of Intermetallic Compounds and Combinations Generally Stayed and Their ReviewHas SimNo ratings yet
- ZetaDocument10 pagesZetaDiana Codreanu SiretanuNo ratings yet
- Roto 2002Document8 pagesRoto 2002k.suganeswaranNo ratings yet
- Mechanism of Corona Treatment On Polyolefin FilmsDocument6 pagesMechanism of Corona Treatment On Polyolefin Filmskavitha ganesanNo ratings yet
- Electrodeposition of Conducting Transition Metal Oxide/Hydroxide Films From Aqueous SolutionDocument4 pagesElectrodeposition of Conducting Transition Metal Oxide/Hydroxide Films From Aqueous SolutionusercmdmcNo ratings yet
- JCIS HysteresisDocument9 pagesJCIS HysteresisShibsekhar RoyNo ratings yet
- Molecular Dynamics of Ionic Transport in Silica ChannelsDocument11 pagesMolecular Dynamics of Ionic Transport in Silica ChannelssaeedNo ratings yet
- Polymer: Jan C. Uecker, Gary C. Tepper, Joan Rosell-LlompartDocument8 pagesPolymer: Jan C. Uecker, Gary C. Tepper, Joan Rosell-LlompartGilbert AnnoheneNo ratings yet
- Magnetically-Induced Flow During ElectropolishingDocument6 pagesMagnetically-Induced Flow During Electropolishingmohammadreza hajialiNo ratings yet
- Capillary Electrophoresis: S. F. Y. Li and Y. S. Wu, National University ofDocument12 pagesCapillary Electrophoresis: S. F. Y. Li and Y. S. Wu, National University ofskljoleNo ratings yet
- The Anodic Dissolution Processes of Copper in Sodium Fluoride SolutionDocument10 pagesThe Anodic Dissolution Processes of Copper in Sodium Fluoride Solution戴海龙No ratings yet
- Conducting Polymers: An IntroductionDocument7 pagesConducting Polymers: An Introductionsjktnknsjgrg100% (1)
- Apparent Inverse Gibbs-Thomson Effect in Dealloyed Nanoporous NanoparticlesDocument5 pagesApparent Inverse Gibbs-Thomson Effect in Dealloyed Nanoporous NanoparticlesParamita HaldarNo ratings yet
- J Jelechem 2006 11 008Document7 pagesJ Jelechem 2006 11 008Mateo bolañosNo ratings yet
- North-Holland Publishing CompanyDocument5 pagesNorth-Holland Publishing CompanyasdfNo ratings yet
- Grain Boundaries in MetalsDocument2 pagesGrain Boundaries in MetalsAvanish KumarNo ratings yet
- Electro Osmosis Theory and ApplicationsDocument15 pagesElectro Osmosis Theory and Applicationskalpna_n3No ratings yet
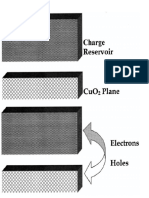
- Electronic Layers in Copper Oxide SuperconductorsDocument1 pageElectronic Layers in Copper Oxide SuperconductorsSJNo ratings yet
- Electrospinning Distribution of Charges in Liquid JetsDocument6 pagesElectrospinning Distribution of Charges in Liquid Jetsxiaobing DongNo ratings yet
- Li DopingDocument5 pagesLi DopingmariaNo ratings yet
- Cicatrices Et Kinésithérapie Après Cancer Du Sein.Document5 pagesCicatrices Et Kinésithérapie Après Cancer Du Sein.Calloch DamienNo ratings yet
- Nanofluidic Dielectrophoresis Single Molecules Holzel PRL 2005Document4 pagesNanofluidic Dielectrophoresis Single Molecules Holzel PRL 2005Vasu ManchesterNo ratings yet
- The Effect of Additives On Zinc ElectrodepositionDocument8 pagesThe Effect of Additives On Zinc ElectrodepositionOlasubomi OmowaNo ratings yet
- Polarization Loop Deformations of An Oxygen Deficient PB (ZR 0.25, Ti 0.75) O 3 Ferroelectric Thin FilmDocument11 pagesPolarization Loop Deformations of An Oxygen Deficient PB (ZR 0.25, Ti 0.75) O 3 Ferroelectric Thin FilmLiviu BadeaNo ratings yet
- 1 s2.0 S0955221912003457 MainDocument18 pages1 s2.0 S0955221912003457 MainSena KulaksızNo ratings yet
- Spectrochimica Acta Part A: Molecular and Biomolecular SpectrosDocument6 pagesSpectrochimica Acta Part A: Molecular and Biomolecular SpectrosOlga Viviana Cardenas LunaNo ratings yet
- 2014 - Charge Transport in Thermally Aged Paperabdelmalik2014Document11 pages2014 - Charge Transport in Thermally Aged Paperabdelmalik2014Viviane CalixtoNo ratings yet
- Highly Confined Ions Store Charge More EfficientlyDocument6 pagesHighly Confined Ions Store Charge More EfficientlyFaysal Rahman SunbirNo ratings yet
- Conduction and Polarization Mechanisms and Trends in DielectricsDocument18 pagesConduction and Polarization Mechanisms and Trends in Dielectricsmarco seguelNo ratings yet
- Lin 2004Document4 pagesLin 2004rahma rahmaNo ratings yet
- Low Cost, High Efficiency Solar Cell Based On Dye-Sensitized Colloidal TiO2 FilmDocument4 pagesLow Cost, High Efficiency Solar Cell Based On Dye-Sensitized Colloidal TiO2 FilmWildan MocholladNo ratings yet
- SWCheong TMO Nat - Mater 2007Document2 pagesSWCheong TMO Nat - Mater 2007karishma sualiheenNo ratings yet
- Electrophoretic Deposition of MaterialsDocument27 pagesElectrophoretic Deposition of Materialshichiku4uNo ratings yet
- Conducting Polymers: 22-2-96 by Colin PrattDocument7 pagesConducting Polymers: 22-2-96 by Colin PrattkshitijscribdNo ratings yet
- The Electrical Double Layer and Its StructureDocument7 pagesThe Electrical Double Layer and Its Structurewks38100% (1)
- Molecule-Independent Electrical Switching in PT Organic Monolayer Ti DevicesDocument4 pagesMolecule-Independent Electrical Switching in PT Organic Monolayer Ti Devicesraymond wellNo ratings yet
- Electrochemical Growth and Characterization of Size-Quantized CdTe Thin FilmsDocument6 pagesElectrochemical Growth and Characterization of Size-Quantized CdTe Thin FilmsEdgar Fabian Pinzon NietoNo ratings yet
- 2012 - Tang - Morphology Effect On Polycrystalline CuDocument6 pages2012 - Tang - Morphology Effect On Polycrystalline CuWasim WasimNo ratings yet
- HYDROMETALLURGICAL PROCESSES NOTESDocument110 pagesHYDROMETALLURGICAL PROCESSES NOTESGodfrey BareNo ratings yet
- 10.1016@b0 12 369397 7@00293 4Document7 pages10.1016@b0 12 369397 7@00293 4Drawing and Artistic DecorationsNo ratings yet
- Synthesis and Optical Properties of Uantum-Size Metal Sulfide Particles in Aqueous SolutionDocument4 pagesSynthesis and Optical Properties of Uantum-Size Metal Sulfide Particles in Aqueous SolutionANGIE PAOLA RODELO PANZANo ratings yet
- Effect of Ion Species On Change in Particle Electrophoresis Caused by ChangeDocument6 pagesEffect of Ion Species On Change in Particle Electrophoresis Caused by ChangeGuillermo Alonso Diaz PachecoNo ratings yet
- 2024 03 13-Three Electrode CVDocument10 pages2024 03 13-Three Electrode CVLong BuiNo ratings yet
- Daniel 1949Document6 pagesDaniel 1949George AcostaNo ratings yet
- Weber1991 PDFDocument17 pagesWeber1991 PDFXyNo ratings yet
- Elec Acta 04Document9 pagesElec Acta 04prathapkumar_1990No ratings yet
- A Study of Factors Affecting On The Zeta Potential of Kaolinite and Quartz PowderDocument9 pagesA Study of Factors Affecting On The Zeta Potential of Kaolinite and Quartz PowderLinaUribeNo ratings yet
- Plasmonic Coupling in Noble Metal NanostructuresDocument12 pagesPlasmonic Coupling in Noble Metal Nanostructurestolasa tamasgenNo ratings yet
- Comparative Study of Nanostructured TiO2, ZnO and Bilayer TiO2/ZnO Dye-Sensitized Solar CellsDocument9 pagesComparative Study of Nanostructured TiO2, ZnO and Bilayer TiO2/ZnO Dye-Sensitized Solar CellsbismuthsunilNo ratings yet
- Recovery of Fine and Ultrafine Mineral Particles by Electroflotation - A ReviewDocument16 pagesRecovery of Fine and Ultrafine Mineral Particles by Electroflotation - A ReviewMaría Pía Arancibia BravoNo ratings yet
- Colloid: StabilityDocument11 pagesColloid: StabilityKHAGESHNo ratings yet
- 2014 Charge Transport in Thermally Aged Paper Impregnated With Natu Ral EsterDocument11 pages2014 Charge Transport in Thermally Aged Paper Impregnated With Natu Ral EsterViviane CalixtoNo ratings yet
- Minerals Engineering: Adrián Rojo, Henrik K. Hansen, Omara MonárdezDocument5 pagesMinerals Engineering: Adrián Rojo, Henrik K. Hansen, Omara MonárdezJHON DENNIS HUAMANVILCA PUMANo ratings yet
- Experimental Design Applied To The Chemical Durability of Sol-Gel-Derived ZirconiasDocument8 pagesExperimental Design Applied To The Chemical Durability of Sol-Gel-Derived ZirconiasMarioNo ratings yet
- Preparation of Zirconia Dental Crowns Via Electrophoretic DepositionDocument8 pagesPreparation of Zirconia Dental Crowns Via Electrophoretic DepositionMarioNo ratings yet
- Analysis of Berkovich Indentation: Pergamon 002IK7683 (95) 00033-XDocument28 pagesAnalysis of Berkovich Indentation: Pergamon 002IK7683 (95) 00033-XMarioNo ratings yet
- Synthetic Graft Substitutes PDFDocument8 pagesSynthetic Graft Substitutes PDFMarioNo ratings yet
- Surface Characterization of Anodically Treated Titanium Alloy For Biomedical ApplicationsDocument8 pagesSurface Characterization of Anodically Treated Titanium Alloy For Biomedical ApplicationsMarioNo ratings yet
- Modeling The Water-Bioglass Interface by Ab Initio Molecular Dynamics SimulationsDocument10 pagesModeling The Water-Bioglass Interface by Ab Initio Molecular Dynamics SimulationsMarioNo ratings yet
- Surface Characterization of Anodically Treated Titanium Alloy For Biomedical ApplicationsDocument8 pagesSurface Characterization of Anodically Treated Titanium Alloy For Biomedical ApplicationsMarioNo ratings yet
- 1 s2.0 S0013468605008200 Main PDFDocument5 pages1 s2.0 S0013468605008200 Main PDFMarioNo ratings yet
- Milos EvDocument27 pagesMilos EvMarioNo ratings yet
- Cracking of Thin Films Bonded To Elastic-Plastic SubstratesDocument18 pagesCracking of Thin Films Bonded To Elastic-Plastic SubstratesMarioNo ratings yet
- EBB 324 Lecture 8Document65 pagesEBB 324 Lecture 8MarioNo ratings yet
- 1 s2.0 S0022309301008225 Main PDFDocument12 pages1 s2.0 S0022309301008225 Main PDFMarioNo ratings yet
- Prospective Observation of CAD/CAM Titanium Ceramic Single Crowns: A Three-Year Follow UpDocument8 pagesProspective Observation of CAD/CAM Titanium Ceramic Single Crowns: A Three-Year Follow UpMarioNo ratings yet
- Wear and tribocorrosion of 304L SS, Zr alloys, and Ti in nitric acidDocument11 pagesWear and tribocorrosion of 304L SS, Zr alloys, and Ti in nitric acidMarioNo ratings yet
- Materials Letters: H.C. Li, D.G. Wang, J.H. Hu, C.Z. ChenDocument4 pagesMaterials Letters: H.C. Li, D.G. Wang, J.H. Hu, C.Z. ChenMarioNo ratings yet
- Electrodeposition of Metals From Non-Aqueous Solutions: Electrochimica ActaDocument13 pagesElectrodeposition of Metals From Non-Aqueous Solutions: Electrochimica ActaMarioNo ratings yet
- Resis A La Fractura en Rec de ZR Sobre TiDocument6 pagesResis A La Fractura en Rec de ZR Sobre TiMarioNo ratings yet
- Electrodeposition of Metals From Non-Aqueous Solutions: Electrochimica ActaDocument13 pagesElectrodeposition of Metals From Non-Aqueous Solutions: Electrochimica ActaMario100% (1)
- 1 s2.0 S014296120100206X MainDocument9 pages1 s2.0 S014296120100206X MainMarioNo ratings yet
- 1 s2.0 S0013794498000526 MainDocument22 pages1 s2.0 S0013794498000526 MainMarioNo ratings yet
- 1 s2.0 S0168874X11000485 Main PDFDocument12 pages1 s2.0 S0168874X11000485 Main PDFMarioNo ratings yet
- Measuring Methods and Applications of NanoindentationDocument17 pagesMeasuring Methods and Applications of NanoindentationMarioNo ratings yet
- 1 s2.0 0013468695004262 MainDocument9 pages1 s2.0 0013468695004262 MainMarioNo ratings yet
- 1 s2.0 S004060900301071X MainDocument7 pages1 s2.0 S004060900301071X MainMarioNo ratings yet
- Corrosion Characterization of Titanium Alloys by Electrochemical TechniquesDocument5 pagesCorrosion Characterization of Titanium Alloys by Electrochemical TechniquesMarioNo ratings yet
- Rec de HA Por Pulverizacion Por PlasmaDocument8 pagesRec de HA Por Pulverizacion Por PlasmaMarioNo ratings yet
- 1 s2.0 S1569190X08000981 MainDocument17 pages1 s2.0 S1569190X08000981 MainMarioNo ratings yet
- Vidrios BioactivosDocument26 pagesVidrios BioactivosMarioNo ratings yet
- Zirconia y Bioglass Ceramica DentalDocument22 pagesZirconia y Bioglass Ceramica DentalMarioNo ratings yet
- Refractory Dry Out Procedures SummaryDocument18 pagesRefractory Dry Out Procedures SummaryRajendran Srn100% (6)
- IDEAL GAS Vs REAL GASDocument3 pagesIDEAL GAS Vs REAL GASJopie ArandaNo ratings yet
- Electromagnetism ReferencesDocument7 pagesElectromagnetism ReferencesNivethithaa DhanrajNo ratings yet
- TK-ALCOZAP-2020-01-06-Rev. DDocument65 pagesTK-ALCOZAP-2020-01-06-Rev. DDaniel Gómez100% (1)
- ModelQuestionsCh16 AKDocument5 pagesModelQuestionsCh16 AKYasmeen ElsawafNo ratings yet
- Calculate Carbonate SystemDocument27 pagesCalculate Carbonate SystemgombossandorNo ratings yet
- Beers Law Lab AP ChemDocument3 pagesBeers Law Lab AP ChembrokenfanNo ratings yet
- Basic Information Needed in Log InterpretationDocument86 pagesBasic Information Needed in Log Interpretationmelannie adanteNo ratings yet
- Semiconductor Assembly ProcessDocument22 pagesSemiconductor Assembly Processlabacahin100% (1)
- Synthesis and Characteristics of Bis (Nitrofurazano) FurazanDocument7 pagesSynthesis and Characteristics of Bis (Nitrofurazano) FurazanBalázs GaramhegyiNo ratings yet
- 472Document35 pages472Nahida BanoNo ratings yet
- Gas Turbine Cycle Calculations and AnalysisDocument3 pagesGas Turbine Cycle Calculations and AnalysisWalter BircherNo ratings yet
- Pipeline JournalDocument68 pagesPipeline Journal구용찬No ratings yet
- A Study On Recent Progress in Electrolyte For Zinc-Air Battery: A Mini ReviewDocument37 pagesA Study On Recent Progress in Electrolyte For Zinc-Air Battery: A Mini Reviewserial dunia odia by sidhuNo ratings yet
- Conduction Through LiquidDocument1 pageConduction Through LiquidMuskan RangrejNo ratings yet
- Curriculum Map Science 7 Fourth QuarterDocument3 pagesCurriculum Map Science 7 Fourth QuarterArze IdleNo ratings yet
- Read The Statement Carefully. Write The Letter of The Correct Answer Before The NumberDocument5 pagesRead The Statement Carefully. Write The Letter of The Correct Answer Before The NumberBeverlyRose Bueno Delos Santos100% (1)
- Design of A Floating Roof Crude Oil Storage Tank of 100,000 BPD Capacity and Prototype FabricationDocument13 pagesDesign of A Floating Roof Crude Oil Storage Tank of 100,000 BPD Capacity and Prototype FabricationMudit VermaNo ratings yet
- RSXYP16 30KJY1 SiE00 07 Part 1 - Service Manuals - EnglishDocument193 pagesRSXYP16 30KJY1 SiE00 07 Part 1 - Service Manuals - EnglishghenceaNo ratings yet
- Water Labs FullDocument4 pagesWater Labs FulljohnosborneNo ratings yet
- Understand Contacts - Midas NFX PDFDocument10 pagesUnderstand Contacts - Midas NFX PDFdzejziNo ratings yet
- Stability Analysis of Soil Slope Subjected To Blast Induced Vibrations Using FLACDocument11 pagesStability Analysis of Soil Slope Subjected To Blast Induced Vibrations Using FLACResmi RosaliniNo ratings yet
- Light - Emitting DiodesDocument12 pagesLight - Emitting DiodesasadNo ratings yet
- A Report On Rail Wheel Interaction and Rail Grinding: Prepared By: Saurabh SharmaDocument15 pagesA Report On Rail Wheel Interaction and Rail Grinding: Prepared By: Saurabh Sharmasemi1919No ratings yet
- OK Tubrod 15.14Document1 pageOK Tubrod 15.14Tiberiu MunteanuNo ratings yet
- Manual Bombas Berkeley PDFDocument24 pagesManual Bombas Berkeley PDFxavifoxNo ratings yet
- Ficha Técnica Activated Carbon Air Filter PDFDocument2 pagesFicha Técnica Activated Carbon Air Filter PDFMeliza SuarezNo ratings yet
- Angular Measurements: MEGR 6181 Engineering MetrologyDocument14 pagesAngular Measurements: MEGR 6181 Engineering MetrologyNarasimha ReddyNo ratings yet
- Rate Data Collection and AnalysisDocument18 pagesRate Data Collection and AnalysisLê MinhNo ratings yet
- Slope Stability Analysis Methods ComparisonDocument143 pagesSlope Stability Analysis Methods ComparisonSachin SharmaNo ratings yet