You might also like
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Revalida 1Document3 pagesRevalida 1herrabiel solisNo ratings yet
- Osce ChecklistDocument11 pagesOsce Checklistgemgem06No ratings yet
- Lecture 5 - Facial Pain and TMJ DiseaseDocument6 pagesLecture 5 - Facial Pain and TMJ DiseaseJeff ChadwickNo ratings yet
- Radiologi Intervensi Di IndiaDocument3 pagesRadiologi Intervensi Di IndiaPutu Adi SusantaNo ratings yet
- Upper-Extremity Deep Venous Thrombosis - ACP HospitalistDocument10 pagesUpper-Extremity Deep Venous Thrombosis - ACP HospitalistSylvia GraceNo ratings yet
- CellCommunication Assessment3Document6 pagesCellCommunication Assessment3Michaela Maria GarciaNo ratings yet
- Heart Reviewer 1Document6 pagesHeart Reviewer 1tokzzNo ratings yet
- Santosh DevDocument45 pagesSantosh Devdevdsantosh100% (1)
- 21 Benign Skin TumorsDocument4 pages21 Benign Skin TumorsAbdul Ghaffar AbdullahNo ratings yet
- Bonus BooksDocument540 pagesBonus BooksDanielle Moore100% (2)
- Netically Modified Crops Their Development, Uses, and Risks by G.H. LiangDocument416 pagesNetically Modified Crops Their Development, Uses, and Risks by G.H. LiangbiokanagasundarNo ratings yet
- 2020 11 07 XII Economics 1Document17 pages2020 11 07 XII Economics 1Sjft6hd FfNo ratings yet
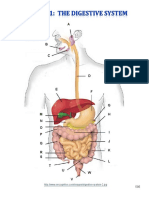
- Digestive System Charts 1Document14 pagesDigestive System Charts 1api-306762527No ratings yet
- Tricks To Remember Important Days-All-MonthsDocument2 pagesTricks To Remember Important Days-All-MonthsSenthikumar MuraliNo ratings yet
- Vestibular SchwannomaDocument12 pagesVestibular SchwannomaEustakia Rini Kartika DewiNo ratings yet
- PNDT Act RulesDocument32 pagesPNDT Act RulesVeerendra BaliNo ratings yet
- Diet & Nutrition Advanced Ver 1Document52 pagesDiet & Nutrition Advanced Ver 1Ina100% (1)
- Ada CambraDocument7 pagesAda CambraCristiano Pereira AlvesNo ratings yet
- Ophtha Quiz - Refractive ErrorsDocument3 pagesOphtha Quiz - Refractive Errorsadi100% (1)
- The Effect of Physiotherapy Intervention by Using Ultrasound and Piriformis Stretching in Management To Reduce Pain in Piriformis SyndromeDocument3 pagesThe Effect of Physiotherapy Intervention by Using Ultrasound and Piriformis Stretching in Management To Reduce Pain in Piriformis SyndromeInternational Journal of Innovative Science and Research TechnologyNo ratings yet
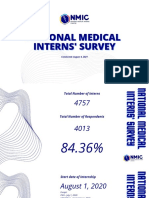
- National Medical National Medical Interns' Survey Interns' SurveyDocument20 pagesNational Medical National Medical Interns' Survey Interns' SurveyKaren Jodes CapananNo ratings yet
- The Sovereign Remedy: Touch-Pieces and The King's Evil. (Pt. I) / Noel WoolfDocument30 pagesThe Sovereign Remedy: Touch-Pieces and The King's Evil. (Pt. I) / Noel WoolfDigital Library Numis (DLN)No ratings yet
- Strategies For Feeding The Preterm Infant: ReviewDocument10 pagesStrategies For Feeding The Preterm Infant: ReviewSanjuy GzzNo ratings yet
- Miner Blood Group SystemDocument44 pagesMiner Blood Group SystemArshad RazaqNo ratings yet
- jfnr06 2 p047 054 Mlichova PDFDocument8 pagesjfnr06 2 p047 054 Mlichova PDFTatum NilamsariNo ratings yet
- Lec 4-Toxicity and Identification TestDocument46 pagesLec 4-Toxicity and Identification TestShoaib MuhammadNo ratings yet
- Effect of Pre-Cooling Injection Site On Pain Perception in Pediatric Dentistry: "A Randomized Clinical Trial"Document6 pagesEffect of Pre-Cooling Injection Site On Pain Perception in Pediatric Dentistry: "A Randomized Clinical Trial"Ummi AzizahNo ratings yet
- Inflammation. Etiology. Vascular Changes. Cellular Events in Inflammation. Acute Inflammation. Morphologic PatternsDocument57 pagesInflammation. Etiology. Vascular Changes. Cellular Events in Inflammation. Acute Inflammation. Morphologic PatternsZauzaNo ratings yet
- 4biomechanics of Removable Partial DenturesDocument15 pages4biomechanics of Removable Partial DenturesDemilyadioppy AbevitNo ratings yet
- Etiology and Diagnosis of Prerenal Disease and Acute Tubular Necrosis in Acute Kidney Injury in Adults - UpToDateDocument22 pagesEtiology and Diagnosis of Prerenal Disease and Acute Tubular Necrosis in Acute Kidney Injury in Adults - UpToDatecuentaparatrabajosdelau10No ratings yet