You might also like
- Tarea Preexamen de Fisicoquímica IDocument15 pagesTarea Preexamen de Fisicoquímica IAlexis Cisneros100% (1)
- Equilibrio Liquido Gas-FisicoquimicaDocument34 pagesEquilibrio Liquido Gas-FisicoquimicaYurika Toledo100% (1)
- Ecuación de Estado de RedlichDocument13 pagesEcuación de Estado de RedlichAlis Carbajal PinedaNo ratings yet
- Guía de Problemas Equilibrio Líquido VaporDocument4 pagesGuía de Problemas Equilibrio Líquido VaporGerson SilvaNo ratings yet
- Problemas Resueltos Del Tema 1Document9 pagesProblemas Resueltos Del Tema 1Ángela Muñoz Chicharro50% (2)
- Práctica Volumen Molar ParcialDocument17 pagesPráctica Volumen Molar ParcialMelissa Salgado100% (1)
- Teoría Del Gas Real: La Relación Exacta Para Gases RealesFrom EverandTeoría Del Gas Real: La Relación Exacta Para Gases RealesNo ratings yet
- Formulario TermodinamicaDocument2 pagesFormulario TermodinamicaRasLyon BlessNo ratings yet
- Conceptos de Fugacidad y ActividadDocument10 pagesConceptos de Fugacidad y ActividadMauro SuZef67% (3)
- Actividad y Coeficiente de ActividadDocument24 pagesActividad y Coeficiente de ActividadTania Flores100% (2)
- Coeficiente de ActividadDocument2 pagesCoeficiente de ActividadMontse CruzNo ratings yet
- Practica 6. Sintesis de Alquenos (Etileno)Document10 pagesPractica 6. Sintesis de Alquenos (Etileno)Javier AyalaNo ratings yet
- La Teroria de Las Velocidades AbsolutasDocument7 pagesLa Teroria de Las Velocidades AbsolutasIsai Keoma Chirinos DiazNo ratings yet
- Actividad y Coeficiente de ActividadDocument4 pagesActividad y Coeficiente de ActividadCrishtian Perez MenesesNo ratings yet
- FISICOQUIMICA2Document14 pagesFISICOQUIMICA2Jesus Alexis Cervantes RamirezNo ratings yet
- Propiedades CríticasDocument5 pagesPropiedades CríticasPaola Carpio75% (4)
- Orta Pérez - PL2 y 3pdfDocument35 pagesOrta Pérez - PL2 y 3pdfOsmyy OrtaNo ratings yet
- 1.5. FisicoquimicaDocument8 pages1.5. FisicoquimicaMith EcheloonNo ratings yet
- Modelos de SolucionDocument5 pagesModelos de SolucionDaniel Gonzales ZayasNo ratings yet
- Cuestiones para Discutir Capitulo 2Document3 pagesCuestiones para Discutir Capitulo 2Fernando Cordova PardoNo ratings yet
- 2.2 Potencial QuimicoDocument3 pages2.2 Potencial QuimicoJulianNacimAsNo ratings yet
- Catálisis Homogénea Por Compuestos de CoordinaciónDocument32 pagesCatálisis Homogénea Por Compuestos de CoordinaciónJohan Delgado100% (1)
- Sistemas No IdealesDocument36 pagesSistemas No IdealesArturo CollazosNo ratings yet
- Ley de Fick2.01Document21 pagesLey de Fick2.01Kristian Moreta0% (1)
- Propiedades Coligativas Ec. MargulesDocument57 pagesPropiedades Coligativas Ec. MargulesricardoNo ratings yet
- Gonzalez Hernandez t1Document9 pagesGonzalez Hernandez t1Veneno Alan Joqsan100% (2)
- Laboratorio 2 ELECTROGRAVIMETRIADocument12 pagesLaboratorio 2 ELECTROGRAVIMETRIAJuan Pablo Squella100% (1)
- Cap 10Document25 pagesCap 10Jessy RamirezNo ratings yet
- Mezcla IdealDocument3 pagesMezcla IdealMiguel Angel Paredes HernándezNo ratings yet
- UNIDAD 5 FLUJO DE EFECTIVO Docx 1Document25 pagesUNIDAD 5 FLUJO DE EFECTIVO Docx 1Thane Botticelli100% (2)
- Balances MolaresDocument7 pagesBalances MolaresAlejandro MerkNo ratings yet
- Potenciales Termodinámicos y Criterios de Espontaneidad y EquilibrioDocument15 pagesPotenciales Termodinámicos y Criterios de Espontaneidad y EquilibrioDaniel Melo100% (1)
- Serie 3 - Equilibrio de Fases - ResueltosDocument5 pagesSerie 3 - Equilibrio de Fases - ResueltosDidier DetchemendyNo ratings yet
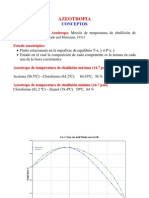
- AzeotropiaDocument25 pagesAzeotropiaRicardo KobainNo ratings yet
- Formulario Basico Fenómenos de Transporte PDFDocument2 pagesFormulario Basico Fenómenos de Transporte PDFCristian SinghNo ratings yet
- Coeficiente de Actividad InformeDocument5 pagesCoeficiente de Actividad InformeJesús Cristhian ChipanaNo ratings yet
- Equilibrio Liquido-Liquido PDFDocument12 pagesEquilibrio Liquido-Liquido PDFAle Jaime0% (1)
- Aplicaciones de Las Propiedades ColigativasDocument14 pagesAplicaciones de Las Propiedades ColigativasAlbert V. EspinozaNo ratings yet
- Difusión Molecular en Gases (1.4)Document5 pagesDifusión Molecular en Gases (1.4)Arlen MamaniNo ratings yet
- Practica 2 SemibatchDocument26 pagesPractica 2 SemibatchJose Manuel Saldaña Ortiz100% (1)
- Metodo Del Calculo de Transferencia de Calor Por Metodo Ntu y DMLTDocument10 pagesMetodo Del Calculo de Transferencia de Calor Por Metodo Ntu y DMLTMiguel CastroNo ratings yet
- Actividad y Coef de Act FISICOQUIMICA IDocument15 pagesActividad y Coef de Act FISICOQUIMICA IOswaldo Rodriguez50% (2)
- Contradifusión Equimolar Parte 3Document6 pagesContradifusión Equimolar Parte 3José GuacollanteNo ratings yet
- Cuestiones para Discutir Capítulo 8Document3 pagesCuestiones para Discutir Capítulo 8Isaac Pineda Carbajal100% (2)
- Propiedades Termodinamicas de Las SolucionesDocument11 pagesPropiedades Termodinamicas de Las SolucionesUlisesYazielSosaLopezNo ratings yet
- Condensadores de Vapor MezcladosDocument3 pagesCondensadores de Vapor MezcladosAnaGomezNo ratings yet
- 06 Problemas Unidad 2Document11 pages06 Problemas Unidad 2Maya MendozaNo ratings yet
- Por Ciento de Conversión, Selectividad y Rendimiento de Una ReacciónDocument12 pagesPor Ciento de Conversión, Selectividad y Rendimiento de Una ReacciónAlexis R AC50% (2)
- Método de Las Velocidades InicialesDocument5 pagesMétodo de Las Velocidades InicialesFitoFuckNo ratings yet
- Potencial Químico de Un Gas Ideal en Una MezclaDocument2 pagesPotencial Químico de Un Gas Ideal en Una MezclaChicho XixiNo ratings yet
- Cuestionario 9 Fenomenos de TransporteDocument6 pagesCuestionario 9 Fenomenos de TransporteOscarMarin100% (1)
- 09-Transferencia de Masa Conveccion Natural y Forzada 2015 Rev2 PDFDocument7 pages09-Transferencia de Masa Conveccion Natural y Forzada 2015 Rev2 PDFRolando RivasNo ratings yet
- Ejercicios para Resolver Diseño ReactorDocument1 pageEjercicios para Resolver Diseño Reactorbeto nuñezNo ratings yet
- La FugacidadDocument5 pagesLa FugacidadMaria Alejandra Narvaez Izaguirre83% (6)
- Ley de Henry y OtrosDocument6 pagesLey de Henry y OtrosCarlos CcqNo ratings yet
- Las Disoluciones Se Forman Cuando Las Fuerzas de Atracción Entre Las PartículasDocument2 pagesLas Disoluciones Se Forman Cuando Las Fuerzas de Atracción Entre Las PartículasDiego MuñozNo ratings yet
- Ley de HenrryDocument7 pagesLey de HenrryJhon ChavezNo ratings yet
- Disoluciones IdealesDocument9 pagesDisoluciones IdealesSteven Hicks100% (2)
- Psicometría. Principios básicos y protocolos experimentales diversosFrom EverandPsicometría. Principios básicos y protocolos experimentales diversosNo ratings yet
- Parra Avila Omar DiegoDocument1 pageParra Avila Omar DiegoOmar Diego ParraNo ratings yet
- NotasDocument2 pagesNotasOmar Diego ParraNo ratings yet
- Cambios en El Rol de La Mujer Desde 1945Document16 pagesCambios en El Rol de La Mujer Desde 1945Omar Diego ParraNo ratings yet
- COMPENDIO 03 - Gestion Ambiental PDFDocument316 pagesCOMPENDIO 03 - Gestion Ambiental PDFOmar Diego ParraNo ratings yet
- Diapositivas Transferencia de Calor 2018-IDocument60 pagesDiapositivas Transferencia de Calor 2018-IOmar Diego ParraNo ratings yet
- Proceso de La CervezaDocument12 pagesProceso de La CervezaOmar Diego ParraNo ratings yet
- Transmision de Calor InformeDocument4 pagesTransmision de Calor InformeCarlos Aldair Velasquez0% (2)
- Previo 9Document2 pagesPrevio 9Polly AsecasNo ratings yet
- Informe CalderaDocument12 pagesInforme CalderaAlberto TovarNo ratings yet
- Linea Del TiempoDocument1 pageLinea Del TiemposergioNo ratings yet
- Ìnstalaciones de VaporDocument95 pagesÌnstalaciones de VaporDavid Arteaga100% (1)
- MARCO TEORICO WordDocument1 pageMARCO TEORICO WordBetoCoronelVallejosNo ratings yet
- Practica de Intercambiadores de Calor de Coraza y TubosDocument24 pagesPractica de Intercambiadores de Calor de Coraza y TubosAlejandro Estrada TorricoNo ratings yet
- FULTONDocument1 pageFULTONclaudia100% (2)
- Bomba CalorimetricaDocument6 pagesBomba CalorimetricaLuis Alejandro Araya OlivaNo ratings yet
- Tarea 2. Carlos HerreraDocument3 pagesTarea 2. Carlos HerreraJavier BaronaNo ratings yet
- Carrier Tarfia Mayo 2023Document164 pagesCarrier Tarfia Mayo 2023rosario peseros castroNo ratings yet
- Energía Libre y Potencial TermodinámicoDocument7 pagesEnergía Libre y Potencial TermodinámicoSari BustillosNo ratings yet
- Guia TDCDocument4 pagesGuia TDCOrnella EseVeNo ratings yet
- Fluidos HoyDocument32 pagesFluidos HoyFernanda GonzalezNo ratings yet
- 7645guía06 1c20.docx-9Document10 pages7645guía06 1c20.docx-9stephanie ilnickiNo ratings yet
- FENÓMENOS CRÍTICOS ccr7Document13 pagesFENÓMENOS CRÍTICOS ccr7Anonymous wH8gUfAFnNo ratings yet
- Carga Térmica CuestionarioDocument1 pageCarga Térmica CuestionarioArzamendiaLGNo ratings yet
- Ciclos de VaporDocument55 pagesCiclos de Vaporsergio baezNo ratings yet
- Ejercicios Resueltos TermodinamicaDocument6 pagesEjercicios Resueltos TermodinamicaJose Luis Martinez Guzman0% (1)
- Fisico Quimica Calor de MezclasDocument3 pagesFisico Quimica Calor de MezclasYandri Javier Sanchez Guerrero100% (1)
- La CondensacionDocument17 pagesLa CondensacionInformatica Municipalidad de ZacapaNo ratings yet
- Fundameto Teorico Presion de VaporDocument5 pagesFundameto Teorico Presion de VaporSmithAnthonyBlasMNo ratings yet
- Calculo de Cargas TermicasDocument12 pagesCalculo de Cargas Termicasomar ruiz lopez100% (1)
- 561 Análisis Exergético A Una Turbina de Gas RegenerativaDocument6 pages561 Análisis Exergético A Una Turbina de Gas RegenerativaHelen LugoNo ratings yet
- Calculo de Tiempo de SecadoDocument22 pagesCalculo de Tiempo de SecadoHalo Prada100% (2)
- AdsorciónDocument23 pagesAdsorciónMaria Fernanda Rached ZamoraNo ratings yet
- Calor Absorbido InformeDocument19 pagesCalor Absorbido InformeJean OrtizNo ratings yet
- Patricia Morales - TAREA 5-FÍSICADocument5 pagesPatricia Morales - TAREA 5-FÍSICAWalter Guzman MezaNo ratings yet
- Guia FenómenosDocument4 pagesGuia FenómenosAarón UlloaNo ratings yet