You might also like
- Cell Biology Exam 1 ReaDocument7 pagesCell Biology Exam 1 ReaCsa At Uh100% (3)
- Questions Bank For Molecular Biology Answer SheetDocument51 pagesQuestions Bank For Molecular Biology Answer Sheetmartynapet85% (39)
- Principles and Applications of Molecular DiagnosticsFrom EverandPrinciples and Applications of Molecular DiagnosticsNader RifaiRating: 5 out of 5 stars5/5 (2)
- Histology NotesDocument4 pagesHistology NotesThonieroce Apryle Jey Morelos100% (2)
- Genetics Problems With AnswersDocument45 pagesGenetics Problems With Answersprofptrajasekharan84% (57)
- Cel Mol LecDocument3 pagesCel Mol LecIzza LimNo ratings yet
- MCQ On Cytology - Cell and Cell Organelles MCQ Biology - Learning Biology Through MCQsDocument5 pagesMCQ On Cytology - Cell and Cell Organelles MCQ Biology - Learning Biology Through MCQsSenthilkumar Palanisamy100% (1)
- Cell Final ExamDocument8 pagesCell Final ExamJorge GomezNo ratings yet
- Essentials of Molecular BiologyDocument770 pagesEssentials of Molecular BiologyTuan Lam Ngoc86% (7)
- Histology: Lecture NotesDocument97 pagesHistology: Lecture Notessky100% (4)
- Fundamentals of the Stem Cell Debate: The Scientific, Religious, Ethical, and Political IssuesFrom EverandFundamentals of the Stem Cell Debate: The Scientific, Religious, Ethical, and Political IssuesRating: 3 out of 5 stars3/5 (1)
- Final Exam Study Guide TipsDocument22 pagesFinal Exam Study Guide Tipsrazgriz1211No ratings yet
- Human Genetics: From DNA to ProteinsDocument28 pagesHuman Genetics: From DNA to ProteinsjasNo ratings yet
- Cellular Molecular BiologyDocument4 pagesCellular Molecular BiologyCpopNo ratings yet
- LUMS Advanced Molecular Biology Course Fall 2018Document4 pagesLUMS Advanced Molecular Biology Course Fall 2018Anonymous sF8ZuiGNo ratings yet
- Basics of Molecular BiologyDocument69 pagesBasics of Molecular BiologypathinfoNo ratings yet
- Molecular Cell Biology Practice QuestionsDocument9 pagesMolecular Cell Biology Practice Questionssahana2791100% (1)
- Medical Genetics 2013 3rdDocument33 pagesMedical Genetics 2013 3rdAbdellatif Elhadad100% (1)
- Genetics WorkbookDocument57 pagesGenetics WorkbookLauren WinnettNo ratings yet
- CH - 40 - Introduction To The Cell CycleDocument15 pagesCH - 40 - Introduction To The Cell Cycleerichaas100% (1)
- Examn of Molecular BiologyDocument11 pagesExamn of Molecular BiologycegfracNo ratings yet
- Molecular GeneticsDocument3 pagesMolecular GeneticsPRINTDESK by DanNo ratings yet
- ImmunologyDocument1 pageImmunologyChris_Barber0986% (7)
- Sorenson Atlas of Human Histology Chapters 1 and 14Document56 pagesSorenson Atlas of Human Histology Chapters 1 and 14Nick john CaminadeNo ratings yet
- Mitosis and MeiosisDocument56 pagesMitosis and MeiosisJaime Soriano100% (1)
- Cell Biology IntroductionDocument1 pageCell Biology IntroductionChris_Barber09100% (1)
- Bacterial MetabolismDocument46 pagesBacterial MetabolismMakarios S. YousefNo ratings yet
- Summary Molecular Biology of The Cell Chapter 17Document9 pagesSummary Molecular Biology of The Cell Chapter 17Yi Jun Hu100% (2)
- Cell Signaling IDocument21 pagesCell Signaling IGuhanNo ratings yet
- Introduction To Molecular BiologyDocument82 pagesIntroduction To Molecular Biologyfarzaana100% (2)
- Histology Laboratory Manual: Olgga A. Hara MSDocument77 pagesHistology Laboratory Manual: Olgga A. Hara MSMark LopezNo ratings yet
- Immunology NotesDocument22 pagesImmunology Notesvyasakandarp100% (3)
- Avi's Notes Clinical BiochemistryDocument171 pagesAvi's Notes Clinical BiochemistryErfaneh FN100% (1)
- Lecture 1 - Introduction To Molecular Biology KMDocument53 pagesLecture 1 - Introduction To Molecular Biology KMcarlNo ratings yet
- Molecular Biology ExamDocument8 pagesMolecular Biology Exam畏No ratings yet
- Histology Summary TableDocument5 pagesHistology Summary TableCec DfNo ratings yet
- Multiple Choice Questions in Molecular BiologyDocument72 pagesMultiple Choice Questions in Molecular Biologydanishuddin_danish50% (2)
- CSIR-NET Cell Biology NotesDocument32 pagesCSIR-NET Cell Biology NotesSrinivas Nakka100% (1)
- Recombinant DNA TechnologyDocument56 pagesRecombinant DNA TechnologyAnkita Priyadarshini100% (1)
- Introduction To Molecular BiologyDocument39 pagesIntroduction To Molecular BiologyAbbinaya100% (9)
- Quizlet GeneticsDocument4 pagesQuizlet GeneticsMa Cecilia SorianoNo ratings yet
- HistologyDocument61 pagesHistologysofy_aboshokNo ratings yet
- Cell Biology Multiple ChoiceDocument47 pagesCell Biology Multiple Choicezuft82% (11)
- Genetic EngineeringDocument218 pagesGenetic EngineeringSrramNo ratings yet
- Regulation of Gene ExpressionDocument20 pagesRegulation of Gene ExpressionrainabtNo ratings yet
- Immunology Lecture 03 - Antibodies Part 1 - 2018Document36 pagesImmunology Lecture 03 - Antibodies Part 1 - 2018api-273068056No ratings yet
- Lecture 1 - Chromosome & GenomeDocument64 pagesLecture 1 - Chromosome & GenomeFriendlyGoodGirlNo ratings yet
- Genome EditingDocument17 pagesGenome EditingdearbhupiNo ratings yet
- BacteriologyDocument75 pagesBacteriologyHoward BarlomentoNo ratings yet
- Kaplan Biochemistry: Study Online atDocument2 pagesKaplan Biochemistry: Study Online atShaz Chindhy100% (1)
- A Text - Book of Histology: Descriptive and Practical. For the Use of StudentsFrom EverandA Text - Book of Histology: Descriptive and Practical. For the Use of StudentsNo ratings yet
- Cell Press Reviews: Core Concepts in Cell BiologyFrom EverandCell Press Reviews: Core Concepts in Cell BiologyCell PressNo ratings yet
- Midwifery Practice TestDocument51 pagesMidwifery Practice TestDennis Nabor Muñoz, RN,RM100% (1)
- Financial Education Report 2021 Part 1.2Document15 pagesFinancial Education Report 2021 Part 1.2Dennis Nabor Muñoz, RN,RMNo ratings yet
- Improve Your Medication Management Processes: Advisory ServicesDocument2 pagesImprove Your Medication Management Processes: Advisory ServicesSimarpreet KaurNo ratings yet
- Care of Clients With Problems Related To The Musculoskeletal SystemDocument46 pagesCare of Clients With Problems Related To The Musculoskeletal SystemMichelle Joy Mallillin MadriaNo ratings yet
- Financial Education Report 2021 Part 1.1Document14 pagesFinancial Education Report 2021 Part 1.1Dennis Nabor Muñoz, RN,RMNo ratings yet
- Covid 19: Dennis N. Muñoz, RN, RM, LPTDocument103 pagesCovid 19: Dennis N. Muñoz, RN, RM, LPTDennis Nabor Muñoz, RN,RMNo ratings yet
- Musculoskeletal Anatomy and Physiology 7Document35 pagesMusculoskeletal Anatomy and Physiology 7Dennis Nabor Muñoz, RN,RMNo ratings yet
- Musculoskeletal Anatomy and Physiology 4Document67 pagesMusculoskeletal Anatomy and Physiology 4Dennis Nabor Muñoz, RN,RM100% (1)
- Musculoskeletal Anatomy and Physiology 8Document41 pagesMusculoskeletal Anatomy and Physiology 8Dennis Nabor Muñoz, RN,RMNo ratings yet
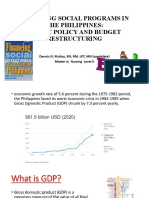
- Financing Social Programs in The Philippines: Public Policy and Budget RestructuringDocument78 pagesFinancing Social Programs in The Philippines: Public Policy and Budget RestructuringDennis Nabor Muñoz, RN,RMNo ratings yet
- Musculoskeletal Anatomy and Physiology 2Document18 pagesMusculoskeletal Anatomy and Physiology 2Dennis Nabor Muñoz, RN,RMNo ratings yet
- Successful StoriesDocument5 pagesSuccessful StoriesDennis Nabor Muñoz, RN,RMNo ratings yet
- Musculoskeletal Anatomy and Physiology 5Document36 pagesMusculoskeletal Anatomy and Physiology 5Dennis Nabor Muñoz, RN,RMNo ratings yet
- Musculoskeletal Anatomy and Physiology 2Document18 pagesMusculoskeletal Anatomy and Physiology 2Dennis Nabor Muñoz, RN,RMNo ratings yet
- Financing Social Programs in The Philippines: Public Policy and Budget RestructuringDocument78 pagesFinancing Social Programs in The Philippines: Public Policy and Budget RestructuringDennis Nabor Muñoz, RN,RMNo ratings yet
- Financial Education Report 2021 Part 1Document28 pagesFinancial Education Report 2021 Part 1Dennis Nabor Muñoz, RN,RMNo ratings yet
- Chapter22 LectureDocument45 pagesChapter22 Lecturecee_prasetyo100% (1)
- Financial Education Report 2021 Part 2Document51 pagesFinancial Education Report 2021 Part 2Dennis Nabor Muñoz, RN,RMNo ratings yet
- Foundations in Microbiology: The Gram-Positive Bacilli of Medical Importance TalaroDocument65 pagesFoundations in Microbiology: The Gram-Positive Bacilli of Medical Importance TalaroOdurNo ratings yet
- The Gram-Negative Bacilli of Medical ImportanceDocument54 pagesThe Gram-Negative Bacilli of Medical ImportanceAna-Maria Nicolae100% (1)
- Chapter23 LectureDocument74 pagesChapter23 Lecture7104No ratings yet
- Foundations in Microbiology: Introduction To Viruses That Infect Humans: The DNA Viruses TalaroDocument50 pagesFoundations in Microbiology: Introduction To Viruses That Infect Humans: The DNA Viruses TalaroDennis Nabor Muñoz, RN,RMNo ratings yet
- Foundations in Microbiology: Miscellaneous Bacterial Agents of Disease TalaroDocument46 pagesFoundations in Microbiology: Miscellaneous Bacterial Agents of Disease TalaroOdurNo ratings yet
- Foundations in Microbiology: The Cocci of Medical Importance TalaroDocument71 pagesFoundations in Microbiology: The Cocci of Medical Importance TalaroOdurNo ratings yet
- Foundations in Microbiology: Disorders in Immunity TalaroDocument43 pagesFoundations in Microbiology: Disorders in Immunity TalaroOdurNo ratings yet
- Foundations in Microbiology: Diagnosing Infections TalaroDocument23 pagesFoundations in Microbiology: Diagnosing Infections TalaroOdurNo ratings yet
- Cell Surface MarkersDocument63 pagesCell Surface MarkersPallavi GhoshNo ratings yet
- Foundations in Microbiology: Microbe-Human Interactions: Infection and Disease TalaroDocument46 pagesFoundations in Microbiology: Microbe-Human Interactions: Infection and Disease TalaroOdurNo ratings yet
- Foundations in Microbiology: Nonspecific Host Defenses TalaroDocument35 pagesFoundations in Microbiology: Nonspecific Host Defenses TalaroOdurNo ratings yet
- Foundations in Microbiology: Drugs, Microbes, Host - The Elements of Chemotherapy TalaroDocument66 pagesFoundations in Microbiology: Drugs, Microbes, Host - The Elements of Chemotherapy TalaroOdurNo ratings yet
- Antibody EngineeringDocument10 pagesAntibody EngineeringSyafrizal MuinNo ratings yet
- Protein Expression and Purification Core Facility - Cloning - Choice of Expression Systems - EMBLDocument4 pagesProtein Expression and Purification Core Facility - Cloning - Choice of Expression Systems - EMBLSanu ShemNo ratings yet
- Protein Synthesis تصنيع البروتينDocument12 pagesProtein Synthesis تصنيع البروتينBra himNo ratings yet
- Invitrogen Price List 2016 (Duty Paid)Document253 pagesInvitrogen Price List 2016 (Duty Paid)Khristin AllisonNo ratings yet
- CELL-PPT-DR.-LINAO, Updated Aug.2,2020Document103 pagesCELL-PPT-DR.-LINAO, Updated Aug.2,2020Allan Q VenusNo ratings yet
- 2962 Plasma Proteins PPT 53b668dea4a12Document35 pages2962 Plasma Proteins PPT 53b668dea4a12mahmoud fuqahaNo ratings yet
- Figure 1 Shows Part of One Strand of A DNA MoleculeDocument12 pagesFigure 1 Shows Part of One Strand of A DNA MoleculeAmal GeorgeNo ratings yet
- DNA Chromatography 2002Document239 pagesDNA Chromatography 2002mehranyNo ratings yet
- HistologyDocument63 pagesHistologyMichelle ThereseNo ratings yet
- Biomolecules crossword puzzleDocument1 pageBiomolecules crossword puzzleSoliel RiegoNo ratings yet
- Cell 101001Document196 pagesCell 101001Prasath100% (1)
- Cell Transport Test ReviewDocument3 pagesCell Transport Test ReviewJohn Kevin NocheNo ratings yet
- Explained - 2020 Nobel Prize in Chemistry - The HinduDocument4 pagesExplained - 2020 Nobel Prize in Chemistry - The HinduIrfan Ullah 116-FBAS/MSCHM/F20No ratings yet
- Mitosis PoemDocument1 pageMitosis PoemParisNo ratings yet
- Kod One PCR Master Mix Kod One PCR Master Mix - Blue-: TM TMDocument15 pagesKod One PCR Master Mix Kod One PCR Master Mix - Blue-: TM TMShima YousefiNo ratings yet
- Real Time PCR: ICMR No. SUPRA001fDocument2 pagesReal Time PCR: ICMR No. SUPRA001fPatel AayushiNo ratings yet
- Hussain 2021Document13 pagesHussain 2021Amín MoraNo ratings yet
- Drugs and The Brain - CourseraDocument5 pagesDrugs and The Brain - CourseraSinisa RisticNo ratings yet
- TT PB 6 Teaching GuideDocument5 pagesTT PB 6 Teaching GuideeashelNo ratings yet
- Christina H. Parks: Magna Cum Laude, Chemistry HonorsDocument2 pagesChristina H. Parks: Magna Cum Laude, Chemistry HonorsTim BrownNo ratings yet
- Materi 5-Bioleaching Mineral OksidaDocument44 pagesMateri 5-Bioleaching Mineral OksidaVicky Faras Barunson PanggabeanNo ratings yet
- Chem 205 Lecture 5: Nucleotides and Nucleic Acids (Derived From Voet, 2011)Document37 pagesChem 205 Lecture 5: Nucleotides and Nucleic Acids (Derived From Voet, 2011)Rab BaloloyNo ratings yet
- 8 2 WKSTDocument3 pages8 2 WKSTapi-262378640No ratings yet
- Topic:: Polymerase Chain Reaction (PCR) Memona Aslam Manisha Mahboob Husnain Javed Iqra Praveen Mam Tehreem 3rd BiologyDocument23 pagesTopic:: Polymerase Chain Reaction (PCR) Memona Aslam Manisha Mahboob Husnain Javed Iqra Praveen Mam Tehreem 3rd BiologyTehreem FatimaNo ratings yet
- Method Used To Demonstrate The Functions of TheDocument24 pagesMethod Used To Demonstrate The Functions of TheRealmeNo ratings yet
- Immune Recap 2Document8 pagesImmune Recap 2ibrahimNo ratings yet
- NATS 1670 Recorded Lecture NotesDocument29 pagesNATS 1670 Recorded Lecture NotescanaliphilipNo ratings yet
- What Are AbzymeDocument2 pagesWhat Are Abzymezakiarbsb100% (1)
- Chemia 3Document1 pageChemia 3juliaNo ratings yet
- 3 - Cellular Respiration NotesDocument22 pages3 - Cellular Respiration Notesapi-375285021No ratings yet