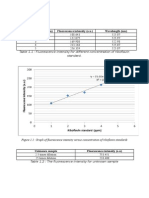
You might also like
- Method Development and Validation of Paracetamol Drug by RP-HPLC 1Document7 pagesMethod Development and Validation of Paracetamol Drug by RP-HPLC 1Anonymous ncDgoMONo ratings yet
- Determination of ParacetamolDocument4 pagesDetermination of ParacetamolChandan SinghNo ratings yet
- Pharmaceutical Analysis of ParacetamolDocument4 pagesPharmaceutical Analysis of ParacetamolFarhana Akter JotyNo ratings yet
- Standard Calibration Curve: PrincipleDocument3 pagesStandard Calibration Curve: PrincipleNeelam MalikNo ratings yet
- GC Lecture NotesDocument8 pagesGC Lecture NotespiyushNo ratings yet
- Simultaneous HPLC Analysis of Betamethasone and Clotrimazole in Cream Formulation PDFDocument4 pagesSimultaneous HPLC Analysis of Betamethasone and Clotrimazole in Cream Formulation PDFNájla KassabNo ratings yet
- Gujarat Technological University: Chemical Technology (36) Subject Code: B.E. 6 SemesterDocument3 pagesGujarat Technological University: Chemical Technology (36) Subject Code: B.E. 6 SemesterDarshanNo ratings yet
- HPLC Method DevelopmentDocument66 pagesHPLC Method DevelopmentPavan Kumar PrathipatiNo ratings yet
- University of Massachusetts Lowell Department of ChemistryDocument68 pagesUniversity of Massachusetts Lowell Department of ChemistrysivabioteckNo ratings yet
- ملزمة رقابة - نسخةDocument68 pagesملزمة رقابة - نسخةتامر الصينيNo ratings yet
- Analytical Chemistry I - Lab Manual - 12102022Document39 pagesAnalytical Chemistry I - Lab Manual - 12102022zahin oktoNo ratings yet
- Lab Manual of Analysis M.pharmDocument37 pagesLab Manual of Analysis M.pharmSridevi G ManipalNo ratings yet
- ZZ1-HPLC 2018-Adsn PDFDocument334 pagesZZ1-HPLC 2018-Adsn PDFSantya Nuhaida LakshitaNo ratings yet
- Book ImpuritiesDocument448 pagesBook ImpuritiesDevang RamoliaNo ratings yet
- Biopharmaceutics and Pharmacokinetics in Drug ResearchDocument20 pagesBiopharmaceutics and Pharmacokinetics in Drug Researchlenanazarova1969No ratings yet
- Method Validation 2012 PDFDocument63 pagesMethod Validation 2012 PDFNam HoaiNo ratings yet
- GMP Project Kuldeep PandeyDocument18 pagesGMP Project Kuldeep PandeyAbhishek JhaNo ratings yet
- Development & Validation of HPLC Analytical Assay For Mefenamic Acid.Document11 pagesDevelopment & Validation of HPLC Analytical Assay For Mefenamic Acid.Anonymous SDUIPeqXNo ratings yet
- Methods of Estimation of MultiDocument41 pagesMethods of Estimation of Multiapi-19786321100% (1)
- Analytical Methods PDFDocument9 pagesAnalytical Methods PDFBabbooNo ratings yet
- Ibuprofen (Pharmacopoea)Document3 pagesIbuprofen (Pharmacopoea)Titis Adisti HapsariNo ratings yet
- Dulal - M.tech (Pharm) - Thesis - Development & Validation - of - HPLC - MethodDocument19 pagesDulal - M.tech (Pharm) - Thesis - Development & Validation - of - HPLC - MethodDulal MahatoNo ratings yet
- Introduction To Pharmaceutical Analysis: AAU, CHS, School of PharmacyDocument75 pagesIntroduction To Pharmaceutical Analysis: AAU, CHS, School of PharmacyBelayneh MathewosNo ratings yet
- Optimization Techniques in Pharmaceutical Formulation KiranDocument22 pagesOptimization Techniques in Pharmaceutical Formulation KiranguslshanNo ratings yet
- HPLC ExperimentDocument4 pagesHPLC ExperimentFrances PaulineNo ratings yet
- A Review On Stability Indicating HPLC Method DevelopmentDocument19 pagesA Review On Stability Indicating HPLC Method DevelopmentppiccoliniNo ratings yet
- Laboratory Manual 20-09-2021Document54 pagesLaboratory Manual 20-09-2021VISHAL ARAVINTH S S (RA2211047010024)No ratings yet
- S.A. Raja Pharmacy College: Vi - Semester - (Iii-B.Pharm)Document51 pagesS.A. Raja Pharmacy College: Vi - Semester - (Iii-B.Pharm)MayurNo ratings yet
- Pre Formulation Stability StudiesDocument33 pagesPre Formulation Stability StudiesDinesh Reddy50% (2)
- Unit 2 - Chemical ReactionsDocument9 pagesUnit 2 - Chemical ReactionsNobukhosi NdlovuNo ratings yet
- GC-MS-MS Analysis of Pesticide Residues in Green Tea Extracted by QuEChERSDocument1 pageGC-MS-MS Analysis of Pesticide Residues in Green Tea Extracted by QuEChERSAmerican Lab100% (1)
- Engineering Mathematics Problems With SolutionsDocument67 pagesEngineering Mathematics Problems With SolutionsCeddi PamiNo ratings yet
- Application of HPLCDocument11 pagesApplication of HPLCIts KazmiNo ratings yet
- Inprocess and Finished Products Quality Control Tests For Pharmaceutical Tablets According To PharmacopoeiasDocument7 pagesInprocess and Finished Products Quality Control Tests For Pharmaceutical Tablets According To PharmacopoeiasMaheshNo ratings yet
- Dissolution Profile ComparisonDocument17 pagesDissolution Profile Comparisondipti_srivNo ratings yet
- Results For Experiment 3Document5 pagesResults For Experiment 3Syahriezan HaminNo ratings yet
- Modern European Pharmacopoeia - Future Trends Cathie VielleDocument53 pagesModern European Pharmacopoeia - Future Trends Cathie Vielledow2008No ratings yet
- Wang Andy Session 21Document45 pagesWang Andy Session 21windli2014No ratings yet
- Calculation and Preparation of Standard Solutions in Food AnalysisDocument5 pagesCalculation and Preparation of Standard Solutions in Food AnalysisMahi HarixNo ratings yet
- Minimum Requirements For Establishment of Pharmacy Institution 17-03-2016Document27 pagesMinimum Requirements For Establishment of Pharmacy Institution 17-03-2016WaqasNo ratings yet
- 1 ThesisDocument236 pages1 ThesisLong ManNo ratings yet
- 8 - Lab8-Potentiometric Titration of Acid MixtureDocument6 pages8 - Lab8-Potentiometric Titration of Acid MixtureHoang Huong TraNo ratings yet
- In Vitro Evaluation of The Pharmaceutical Equivalence of Phenoxymethylpenicillin Tablet Formulations Available in BangladeshDocument3 pagesIn Vitro Evaluation of The Pharmaceutical Equivalence of Phenoxymethylpenicillin Tablet Formulations Available in BangladeshApurba Sarker ApuNo ratings yet
- Dissolution Procedure Test USPDocument16 pagesDissolution Procedure Test USPJorge Estuardo BatzinNo ratings yet
- Sigma Aldrich Grading ChartDocument2 pagesSigma Aldrich Grading Chartjm06100% (1)
- Advanced Pharmaceutical AnalysisDocument4 pagesAdvanced Pharmaceutical AnalysisRezaul RazibNo ratings yet
- 논문 - A stability-indicating HPLC method for the determination of glucosamine in pharmaceutical formulationsDocument7 pages논문 - A stability-indicating HPLC method for the determination of glucosamine in pharmaceutical formulationsjs_kim5781No ratings yet
- Iron Experiment XWDocument4 pagesIron Experiment XWpathisharmaNo ratings yet
- Topical Dermatologic Products - QBDDocument31 pagesTopical Dermatologic Products - QBDvg_vvgNo ratings yet
- Lab 4 HPLCDocument18 pagesLab 4 HPLCDNav14No ratings yet
- Development MethodDocument5 pagesDevelopment MethodBayu RefindraNo ratings yet
- Questions On ISE Choose The Correct AnswerDocument3 pagesQuestions On ISE Choose The Correct Answerنيرمين احمدNo ratings yet
- Chemometrics in EXCEL - 8Document24 pagesChemometrics in EXCEL - 8MAGU_MWENYEWENo ratings yet
- European Journal of Biomedical AND Pharmaceutical SciencesDocument14 pagesEuropean Journal of Biomedical AND Pharmaceutical SciencesSACHIN BHASKAR NARKHEDENo ratings yet
- BP & USP Monographs of IsoniazidDocument11 pagesBP & USP Monographs of IsoniazidRaj GuptaNo ratings yet
- Section-IN PROCESS QUALITY CONTROL and QUALITY CONTROL PDFDocument73 pagesSection-IN PROCESS QUALITY CONTROL and QUALITY CONTROL PDFMd Hasnat Jaman100% (1)
- Essays on Analytical Chemistry: In Memory of Professor Anders RingbomFrom EverandEssays on Analytical Chemistry: In Memory of Professor Anders RingbomErkki WänninenNo ratings yet
- Hasil Praktikum BFFKDocument9 pagesHasil Praktikum BFFKRizqita Atikah SNo ratings yet
- Vol. 3, No. 1, Pebruari 2010 Veterinaria MedikaDocument8 pagesVol. 3, No. 1, Pebruari 2010 Veterinaria MedikaAnonymous SXVNjMcNo ratings yet
- Jurnal Milnacipran HindawiDocument7 pagesJurnal Milnacipran HindawiRizqita Atikah SNo ratings yet
- Cancer Gene TherapyDocument37 pagesCancer Gene TherapyRizqita Atikah SNo ratings yet
- Jurnal 11939 PDFDocument13 pagesJurnal 11939 PDFRizqita Atikah SNo ratings yet
- Hydro GelDocument7 pagesHydro GelRizqita Atikah SNo ratings yet
- I Write A PoemDocument1 pageI Write A PoemRizqita Atikah SNo ratings yet
- Assignment MTO 2 - Unit 1, Unit 2 and Part of Unit 3Document4 pagesAssignment MTO 2 - Unit 1, Unit 2 and Part of Unit 3Shane MandarinNo ratings yet
- A Seminar On A Seminar On: HPLC Detectors HPLC DetectorsDocument36 pagesA Seminar On A Seminar On: HPLC Detectors HPLC DetectorsVivek SagarNo ratings yet
- Chapter 3: Fundamentals of Crystallography: Issues To Address..Document68 pagesChapter 3: Fundamentals of Crystallography: Issues To Address..Jigoku KuroakaNo ratings yet
- Early Theories of Acids and BasesDocument12 pagesEarly Theories of Acids and Basesapi-242798587No ratings yet
- Instrumentation-562-IV-CORE-32511410 - OC-ANALYTICAL INSTRUMENTATION-21 Jun 2021 PDFDocument2 pagesInstrumentation-562-IV-CORE-32511410 - OC-ANALYTICAL INSTRUMENTATION-21 Jun 2021 PDFRicky RawNo ratings yet
- Keto Prof enDocument2 pagesKeto Prof enpipisoseticaNo ratings yet
- Identification and Quantification of Avonoids of Mexican Oregano (Lippia Graveolens) by LC-DAD-ESI/MS AnalysisDocument9 pagesIdentification and Quantification of Avonoids of Mexican Oregano (Lippia Graveolens) by LC-DAD-ESI/MS AnalysisAndrés F. CáceresNo ratings yet
- Buffers Michaels CalculationDocument5 pagesBuffers Michaels CalculationHassan Haider100% (1)
- ASTM E17Document3 pagesASTM E17wendeltrentoNo ratings yet
- Zetium: Elemental ExcellenceDocument9 pagesZetium: Elemental ExcellenceĐức BkNo ratings yet
- 2008 A Levels P1 (No Worked Soln) and P2Document6 pages2008 A Levels P1 (No Worked Soln) and P2toh tim lamNo ratings yet
- Msc-Chemistry (Under CBCS) FIRST YEAR 2017-18 Two Semesters: 15 Weeks EachDocument37 pagesMsc-Chemistry (Under CBCS) FIRST YEAR 2017-18 Two Semesters: 15 Weeks EachHimanshiNo ratings yet
- Mcqs and Solved Short Questions Applied ChemistryDocument25 pagesMcqs and Solved Short Questions Applied ChemistryShahbaz Ahmed RanaNo ratings yet
- Membrane Technology: Ultrafiltration For A Swimming PoolDocument9 pagesMembrane Technology: Ultrafiltration For A Swimming PoolsacNo ratings yet
- Smoker EquationDocument4 pagesSmoker EquationjuanNo ratings yet
- Chemistry Assignment Ayush SharmaDocument14 pagesChemistry Assignment Ayush SharmaAyush SharmaNo ratings yet
- Chemistry Viva QuestionsDocument6 pagesChemistry Viva Questionsandrea234195% (19)
- AcidinjuiceandsodalabrubricDocument11 pagesAcidinjuiceandsodalabrubricapi-302408024No ratings yet
- Kvetoslav R Spurny Editor Analytical Chemistry of Aerosols ScienceDocument499 pagesKvetoslav R Spurny Editor Analytical Chemistry of Aerosols ScienceShibu Arjunan100% (1)
- Dna Extraction From Live Organism Background of The ExperimentDocument2 pagesDna Extraction From Live Organism Background of The Experimentborna goanelNo ratings yet
- Ms-Dial Tsugawa2015Document9 pagesMs-Dial Tsugawa2015delvigoandresNo ratings yet
- Metode Locoparatusin SyrupDocument9 pagesMetode Locoparatusin SyrupHage NdoNo ratings yet
- Nyb U4 Acids Bases Part 1Document67 pagesNyb U4 Acids Bases Part 1Aindrila KaziNo ratings yet
- Eq WT of Salicyclic AcidDocument4 pagesEq WT of Salicyclic AcidRaj Bhan100% (1)
- Solubility Product WorksheetDocument5 pagesSolubility Product WorksheetRyanNo ratings yet
- PJ - 2 - 5 - Bougainvillea Glabra.....Document4 pagesPJ - 2 - 5 - Bougainvillea Glabra.....irishjamile dulosNo ratings yet
- Topic 5 (Updated) Gravimetric Methods of AnalysisDocument38 pagesTopic 5 (Updated) Gravimetric Methods of AnalysisAdznaira AmilussinNo ratings yet
- Physical Chemistry: Target: Jee Main and Advanced 2022Document64 pagesPhysical Chemistry: Target: Jee Main and Advanced 2022sarvesh goyalNo ratings yet
- Unit 7 - Equilibrium NotesDocument174 pagesUnit 7 - Equilibrium NotesRahul Ghosh100% (1)
- (Analytical Chemistry) Acid - Base Titration Lab ReportDocument4 pages(Analytical Chemistry) Acid - Base Titration Lab ReportThu Hien VuNo ratings yet