You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- S-Formula True - StoryDocument550 pagesS-Formula True - StoryAbhishek100% (5)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Reaction Paper On Son RiseDocument3 pagesReaction Paper On Son RiseCarlo P. Carlon71% (14)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Varicose Vein and Naturopathy Treatment Option - A Case ReportDocument5 pagesVaricose Vein and Naturopathy Treatment Option - A Case ReportIJAR JOURNALNo ratings yet
- Method Statement - Fire Rated Duct Insulation.Document16 pagesMethod Statement - Fire Rated Duct Insulation.suhail kalody100% (2)
- Single Parent Households and The Effect On Student Learning PDFDocument69 pagesSingle Parent Households and The Effect On Student Learning PDFMalika AbdullahNo ratings yet
- 3.1 Pre-Delivery and Storage Preparation 3.2 Incoming Storage at SiteDocument4 pages3.1 Pre-Delivery and Storage Preparation 3.2 Incoming Storage at SiteAnis NabilaNo ratings yet
- Case: People vs. Domingo G.R. No.: 184343 Date: March 2, 2009Document2 pagesCase: People vs. Domingo G.R. No.: 184343 Date: March 2, 2009Joyleen HebronNo ratings yet
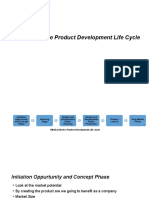
- Medical Device Product Development Life CycleDocument8 pagesMedical Device Product Development Life CycleSuresh Anand100% (1)
- Mop PsoDocument46 pagesMop Psojaved765No ratings yet
- A General Framework For Graph SparsificationDocument10 pagesA General Framework For Graph SparsificationvliviuNo ratings yet
- A Multiobjective Genetic Algorithm To Find Communities in Complex NetworksDocument13 pagesA Multiobjective Genetic Algorithm To Find Communities in Complex NetworksvliviuNo ratings yet
- Level Set Methods in An EM Framework For Shape Classification and EstimationDocument9 pagesLevel Set Methods in An EM Framework For Shape Classification and EstimationvliviuNo ratings yet
- Sparse Nonnegative Solution of Underdetermined Linear Equations by Linear ProgrammingDocument17 pagesSparse Nonnegative Solution of Underdetermined Linear Equations by Linear ProgrammingvliviuNo ratings yet
- Presentation Edulearn2015Document12 pagesPresentation Edulearn2015vliviuNo ratings yet
- Goda Morphological Determination of Pathological PCG Signals by Time and Frequency Domain AnalysisDocument4 pagesGoda Morphological Determination of Pathological PCG Signals by Time and Frequency Domain AnalysisvliviuNo ratings yet
- Generalize DR BFDocument12 pagesGeneralize DR BFvliviuNo ratings yet
- Terabyte-Scale Deep Multiple Instance Learning For Classification and Localization in PathologyDocument18 pagesTerabyte-Scale Deep Multiple Instance Learning For Classification and Localization in PathologyvliviuNo ratings yet
- Extensions of Compressed Sensing: Yaakov Tsaig David L. Donoho October 22, 2004Document20 pagesExtensions of Compressed Sensing: Yaakov Tsaig David L. Donoho October 22, 2004vliviuNo ratings yet
- Neural Network Approaches For Children'S Emotion Recognition in Intelligent Learning ApplicationsDocument12 pagesNeural Network Approaches For Children'S Emotion Recognition in Intelligent Learning ApplicationsvliviuNo ratings yet
- Versiune Finala in Proceedings Id941Document10 pagesVersiune Finala in Proceedings Id941vliviuNo ratings yet
- Wseas09 VladutuDocument6 pagesWseas09 VladutuvliviuNo ratings yet
- Neural Networks Approaches For Children's Emotion Recognition in Inteligent Learning Applications 887 PDFDocument11 pagesNeural Networks Approaches For Children's Emotion Recognition in Inteligent Learning Applications 887 PDFvliviuNo ratings yet
- Versiune Finala in Proceedings Id941Document10 pagesVersiune Finala in Proceedings Id941vliviuNo ratings yet
- Versiune Finala in Proceedings Id941Document10 pagesVersiune Finala in Proceedings Id941vliviuNo ratings yet
- The Supervised Network Self-Organizing Map For Classification of Large Data SetsDocument19 pagesThe Supervised Network Self-Organizing Map For Classification of Large Data SetsvliviuNo ratings yet
- Wseas TransOnSystemsDocument10 pagesWseas TransOnSystemsvliviuNo ratings yet
- JDK1.5 ConcurrentDocument36 pagesJDK1.5 ConcurrentvliviuNo ratings yet
- Parallel Vs DistributedDocument18 pagesParallel Vs DistributedvliviuNo ratings yet
- Enabling Security Essentials For CIOsDocument4 pagesEnabling Security Essentials For CIOsvliviuNo ratings yet
- Online Neural Network Training For Automatic Ischemia Episode DetectionDocument7 pagesOnline Neural Network Training For Automatic Ischemia Episode DetectionvliviuNo ratings yet
- MBEC Final SubmissionDocument24 pagesMBEC Final SubmissionvliviuNo ratings yet
- Models of Computation and Time Complexity: August 3, 2007Document4 pagesModels of Computation and Time Complexity: August 3, 2007vliviuNo ratings yet
- 02 06reportDocument8 pages02 06reportvliviuNo ratings yet
- Adaptive Stepsize Alg 4online TrainingDocument6 pagesAdaptive Stepsize Alg 4online TrainingvliviuNo ratings yet
- Adaptive Stepsize Alg 4online TrainingDocument6 pagesAdaptive Stepsize Alg 4online TrainingvliviuNo ratings yet
- Submission EMBC1Document4 pagesSubmission EMBC1vliviuNo ratings yet
- Bioinf Net SOMDocument22 pagesBioinf Net SOMvliviuNo ratings yet
- PALS Assessment Quiz AnswersDocument3 pagesPALS Assessment Quiz AnswersRea CantoNo ratings yet
- Reflection Paper1 Psy111-1 A41Document1 pageReflection Paper1 Psy111-1 A41owyyyNo ratings yet
- JSA Form - DemolitionDocument5 pagesJSA Form - DemolitionARMANINo ratings yet
- Assessment of Haematological Parameter and Liver Enzyme Among Hepatitis B Infected Blood DonorsDocument9 pagesAssessment of Haematological Parameter and Liver Enzyme Among Hepatitis B Infected Blood DonorsKIU PUBLICATION AND EXTENSIONNo ratings yet
- 12) Types of Clasp AssembliesDocument16 pages12) Types of Clasp Assembliesطيبة نعمانNo ratings yet
- Group 1 Ab12a4 Proposal Marketing PT 1 3Document13 pagesGroup 1 Ab12a4 Proposal Marketing PT 1 3쥬얼이No ratings yet
- H 046 000181 00 3.0 - DPM4 - Service - ManualDocument88 pagesH 046 000181 00 3.0 - DPM4 - Service - ManualZeljko TomicNo ratings yet
- Hipertensi Di PenerbanganDocument3 pagesHipertensi Di PenerbanganEri YunianNo ratings yet
- Sample Patient ProfileDocument7 pagesSample Patient ProfileElisabeth CampbellNo ratings yet
- PR2 Mod 1Document60 pagesPR2 Mod 1Jan Loven Del RosarioNo ratings yet
- Template School ProfileDocument4 pagesTemplate School ProfileCaitlin PeñeraNo ratings yet
- Livro HISTERO VerbookpdfDocument128 pagesLivro HISTERO VerbookpdfMarcus MenezesNo ratings yet
- Pubpol 511Document12 pagesPubpol 511Joshua WhiteNo ratings yet
- 1393un 2023-03Document94 pages1393un 2023-03Jorjen Bryan LeeNo ratings yet
- Biology: Cambridge International Examinations International General Certificate of Secondary EducationDocument20 pagesBiology: Cambridge International Examinations International General Certificate of Secondary Educationmath magicNo ratings yet
- Taski r1 MsdsDocument4 pagesTaski r1 MsdsHemant Sawant50% (10)
- Monitoring school safety at block levelDocument4 pagesMonitoring school safety at block levelSHIVAM KUMARNo ratings yet
- Sitting Kills, Moving Heals. An Interview With Dr. Joan VernikosDocument25 pagesSitting Kills, Moving Heals. An Interview With Dr. Joan Vernikosjayjonbeach100% (1)
- Preoperative Health Status Evaluation: An Accurate Medical HistoryDocument18 pagesPreoperative Health Status Evaluation: An Accurate Medical HistoryNisaNo ratings yet
- Chapter Three The Components of Child Protection: A Brief Overview of Specific Child Protection IssuesDocument10 pagesChapter Three The Components of Child Protection: A Brief Overview of Specific Child Protection IssuesButa DenisNo ratings yet
- Allegation of Rape Cover Up at House of Hope AtlantaDocument2 pagesAllegation of Rape Cover Up at House of Hope AtlantaLeonardo BlairNo ratings yet
- Customer DatabaseDocument78 pagesCustomer DatabaseAnurag SinghNo ratings yet