You might also like
- Rating Rubrics for Applicant EvaluationDocument1 pageRating Rubrics for Applicant EvaluationMike EbarleNo ratings yet
- CompetencyDocument1 pageCompetencyMike EbarleNo ratings yet
- THIS WAY Inverted 12 Inches by 32 Inches (1 Copy)Document1 pageTHIS WAY Inverted 12 Inches by 32 Inches (1 Copy)Mike EbarleNo ratings yet
- HypocalcemiaDocument12 pagesHypocalcemiaKrinessa May100% (1)
- The Presentations of Patients With Hypocalcemia Vary WidelyDocument1 pageThe Presentations of Patients With Hypocalcemia Vary WidelyMike EbarleNo ratings yet
- What Pro Should Inform Ihcps Frequently Asked Questions (Faqs) Health Information Technology Providers (Hitp)Document2 pagesWhat Pro Should Inform Ihcps Frequently Asked Questions (Faqs) Health Information Technology Providers (Hitp)Mike EbarleNo ratings yet
- Bill of Rights2Document7 pagesBill of Rights2Lorigen M. PaternoNo ratings yet
- Emergency CartDocument2 pagesEmergency CartMike EbarleNo ratings yet
- Jaundice InformationDocument4 pagesJaundice InformationMike EbarleNo ratings yet
- Personality Disorder - Cluster C: Anxious and Fearful BehaviorsDocument9 pagesPersonality Disorder - Cluster C: Anxious and Fearful BehaviorsMike EbarleNo ratings yet
- Journal MaternalDocument1 pageJournal MaternalMike EbarleNo ratings yet
- Statistical ModelDocument1 pageStatistical ModelMike EbarleNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5784)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (890)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (72)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Rems FM Shelf May 2022Document80 pagesRems FM Shelf May 2022Manthan PatelNo ratings yet
- Lista BibliográficaDocument17 pagesLista BibliográficafernandaNo ratings yet
- Beyond the Brain: The Systemic Pathophysiological Response to Acute Ischemic StrokeDocument16 pagesBeyond the Brain: The Systemic Pathophysiological Response to Acute Ischemic StrokeMarsya Yulinesia LoppiesNo ratings yet
- Pathology of The Male GUTDocument108 pagesPathology of The Male GUTHoque Mohammed Newaz ShorifulNo ratings yet
- Instant Download Macroeconomics 4th Edition Hubbard Test Bank PDF Full ChapterDocument32 pagesInstant Download Macroeconomics 4th Edition Hubbard Test Bank PDF Full ChapterHowardZunigaxgqt100% (5)
- Choice of Therapy in PrimaryDocument19 pagesChoice of Therapy in PrimaryThiago SartiNo ratings yet
- Note-Making: by Mohua Kundu PGT, English K.V.No-1, Afs, KalaikundaDocument27 pagesNote-Making: by Mohua Kundu PGT, English K.V.No-1, Afs, KalaikundaShady WeebNo ratings yet
- Join Radiotherapy Physics TeamDocument2 pagesJoin Radiotherapy Physics TeamAlina RogojanuNo ratings yet
- Risk Factors for Venous Disease Severity Including Age, Family History & LifestyleDocument1 pageRisk Factors for Venous Disease Severity Including Age, Family History & LifestyleMiguel MendozaNo ratings yet
- Erythrocyte Dysplasia in Peripheral Blood Smears From 5 Thrombocytopenic Dogs Treated With Vincristine SulfateDocument7 pagesErythrocyte Dysplasia in Peripheral Blood Smears From 5 Thrombocytopenic Dogs Treated With Vincristine SulfateLaboratorio de BioquimicaNo ratings yet
- Hematology: - The Science Dealing With The FormationDocument104 pagesHematology: - The Science Dealing With The FormationYamSomandarNo ratings yet
- Nursing Stomach NotesDocument5 pagesNursing Stomach Noteslucas dibenedettoNo ratings yet
- Role of Prostaglandin Analogs in The Treatment of GlaucomaDocument53 pagesRole of Prostaglandin Analogs in The Treatment of GlaucomaAndi Ayu LestariNo ratings yet
- Praktikum Patologi Ikan 2020Document33 pagesPraktikum Patologi Ikan 2020Syifa ekhiasanNo ratings yet
- Case #6 Registered Dental Hygienist (RDH) Magazine PDFDocument1 pageCase #6 Registered Dental Hygienist (RDH) Magazine PDFAlex DaniciNo ratings yet
- Clinical Value of Lead aVRDocument8 pagesClinical Value of Lead aVRmhelguera1No ratings yet
- Conversation of Nurse With PatientDocument2 pagesConversation of Nurse With PatientYola NoviyanaNo ratings yet
- FAMILY NURSING CARE PLAN FOR THE DALIPE FAMILYDocument22 pagesFAMILY NURSING CARE PLAN FOR THE DALIPE FAMILYAdrienne Nicole PaneloNo ratings yet
- Pre-Intermediate Unit 8 VocabularyDocument6 pagesPre-Intermediate Unit 8 VocabularyHama CheikhNo ratings yet
- BSC Nursing SyllabusDocument53 pagesBSC Nursing SyllabussantoshjosephNo ratings yet
- Toxicology Coverage Midterm Exam PDFDocument17 pagesToxicology Coverage Midterm Exam PDFNestor BargioNo ratings yet
- Work Immersion 2023Document12 pagesWork Immersion 2023Cuasay, Vernelle Stephanie De guzmanNo ratings yet
- Fluid and ElectrolytesDocument179 pagesFluid and ElectrolytesTrixie Al Marie75% (4)
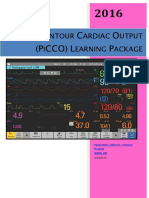
- Pulse Contour Cardiac Output Learning Package PDFDocument17 pagesPulse Contour Cardiac Output Learning Package PDFNurse PractitionerNo ratings yet
- Hospital Acquired InfectionsDocument51 pagesHospital Acquired Infectionstummalapalli venkateswara raoNo ratings yet
- Pogs 2021 Souvenir ProgramDocument264 pagesPogs 2021 Souvenir Programmaria angela SaldajenoNo ratings yet
- Kreozan PoisoningDocument9 pagesKreozan Poisoningdr Alex stanNo ratings yet
- Israeli Scientists Claim To Reverse Aging ProcessDocument2 pagesIsraeli Scientists Claim To Reverse Aging ProcessKarina PolikarpovaNo ratings yet
- High-Sensitivity Cardiac Troponin at Presentation To Rule Out Myocardial Infarction (Historic)Document20 pagesHigh-Sensitivity Cardiac Troponin at Presentation To Rule Out Myocardial Infarction (Historic)DenisseRangelNo ratings yet
- Sas 11 LectureDocument3 pagesSas 11 LectureFretchel Grace Silverado MesaNo ratings yet