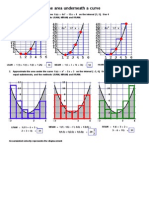
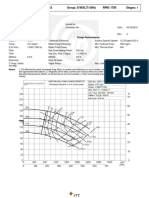
You might also like
- Modeling the Fate and Effect of the Toxic Substances in the EnvironmentFrom EverandModeling the Fate and Effect of the Toxic Substances in the EnvironmentNo ratings yet
- A Systematic Study of Thermochromic Aromatic Donor - Acceptor MaterialsDocument9 pagesA Systematic Study of Thermochromic Aromatic Donor - Acceptor MaterialsDiogomussumNo ratings yet
- Organic Dyes Containing Coplanar Diphenyl-Substituted Dithienosilole Core For Efficient Dye-Sensitized Solar CellsDocument8 pagesOrganic Dyes Containing Coplanar Diphenyl-Substituted Dithienosilole Core For Efficient Dye-Sensitized Solar CellsDiogomussumNo ratings yet
- Gallic 2 AbtDocument7 pagesGallic 2 AbtSiddhesh Umesh MestryNo ratings yet
- Contribution From The Department of Chemistry, University of Waterloo, Waterloo, Ontario N2L 3G1, CanadaDocument12 pagesContribution From The Department of Chemistry, University of Waterloo, Waterloo, Ontario N2L 3G1, CanadaTim TimntimnNo ratings yet
- Ic0614216Document8 pagesIc0614216Rodrigo MartinezNo ratings yet
- Inorg References - Jewel - Arbie - Aya - Roxanne - Amin - Amoloria - Klarize - Jarissa ReferencesDocument19 pagesInorg References - Jewel - Arbie - Aya - Roxanne - Amin - Amoloria - Klarize - Jarissa ReferencesJARISSA DIZON GALLAZANo ratings yet
- Sina Yeganeh, Michael Galperin, and Mark A. Ratner - Switching in Molecular Transport Junctions: Polarization ResponseDocument8 pagesSina Yeganeh, Michael Galperin, and Mark A. Ratner - Switching in Molecular Transport Junctions: Polarization ResponseHumdsNo ratings yet
- Spatiotemporal Microbial Evolution On Antibiotic Landscapes: 13 ReferencesandnotesDocument6 pagesSpatiotemporal Microbial Evolution On Antibiotic Landscapes: 13 ReferencesandnotesJaqueline Godinez CamachoNo ratings yet
- Bauer 2008Document9 pagesBauer 2008Huấn BùiNo ratings yet
- Masato Nozawa Et Al - Total Synthesis of The Hallucinogenic Neoclerodane Diterpenoid Salvinorin ADocument5 pagesMasato Nozawa Et Al - Total Synthesis of The Hallucinogenic Neoclerodane Diterpenoid Salvinorin ABic0000No ratings yet
- CatalysisDocument13 pagesCatalysisSammer BurgosNo ratings yet
- Ultrathin Coatings From Isocyanate-Terminated Star PEGDocument9 pagesUltrathin Coatings From Isocyanate-Terminated Star PEGGanesh KshirsagarNo ratings yet
- Multipolymer Solution-Phase Reactions: Application To The Mitsunobu ReactionDocument2 pagesMultipolymer Solution-Phase Reactions: Application To The Mitsunobu ReactionthamtusieuquayNo ratings yet
- Additions and Corrections: Bhadbhade and D. Srinivas?Document9 pagesAdditions and Corrections: Bhadbhade and D. Srinivas?Lazar AlinaNo ratings yet
- Pacheco Morphology of Aggregatedasphaltene Models 2004Document11 pagesPacheco Morphology of Aggregatedasphaltene Models 2004Ivan Tobar ArteagaNo ratings yet
- Electrophile Affinity: A Reactivity Measure For Aromatic SubstitutionDocument6 pagesElectrophile Affinity: A Reactivity Measure For Aromatic SubstitutionMauricio SánchezNo ratings yet
- Kohn Sham OrbitalsDocument7 pagesKohn Sham OrbitalsAnanya SNo ratings yet
- Josh Vura-Weis Et Al - Geometry and Electronic Coupling in Perylenediimide Stacks: Mapping Structure-Charge Transport RelationshipsDocument2 pagesJosh Vura-Weis Et Al - Geometry and Electronic Coupling in Perylenediimide Stacks: Mapping Structure-Charge Transport RelationshipsGomsajNo ratings yet
- Inorg. Chem.-2010-49-15 - 7007-7015Document9 pagesInorg. Chem.-2010-49-15 - 7007-7015Suman TripathyNo ratings yet
- Spectroscopic Characterization of Spin-Labeled Magnetically Oriented Phospholipid Bilayers by EPR SpectrosDocument7 pagesSpectroscopic Characterization of Spin-Labeled Magnetically Oriented Phospholipid Bilayers by EPR SpectrosSveti JeronimNo ratings yet
- Thermally Stable Single-Atom Platinum-On-Ceria Catalysts Via Atom TrappingDocument6 pagesThermally Stable Single-Atom Platinum-On-Ceria Catalysts Via Atom Trappingsergioodin4851No ratings yet
- Fluoride: Solution-And Solid-State Structural Binding ProbeDocument7 pagesFluoride: Solution-And Solid-State Structural Binding ProbeSilvanaMedhatNo ratings yet
- Titanium and Zirconium Complexes For Polymerization of Propylene and Cyclic EstersDocument11 pagesTitanium and Zirconium Complexes For Polymerization of Propylene and Cyclic EstersTwinkle-Twinkle Little ViezhaNo ratings yet
- Synthesis, Self-Assembly, and Charge Transporting Property of Contorted TetrabenzocoronenesDocument9 pagesSynthesis, Self-Assembly, and Charge Transporting Property of Contorted TetrabenzocoronenesDiogo DiasNo ratings yet
- Joc - 7085Document7 pagesJoc - 7085Diogo DiasNo ratings yet
- Biomimetic Sensor For Certain Phenols Employing A Copper (II) ComplexDocument10 pagesBiomimetic Sensor For Certain Phenols Employing A Copper (II) ComplexAlexandra AlexyaNo ratings yet
- Geoffrey R. Hutchison Et Al - Intermolecular Charge Transfer Between Heterocyclic Oligomers. Effects of Heteroatom and Molecular Packing On Hopping Transport in Organic SemiconductorsDocument16 pagesGeoffrey R. Hutchison Et Al - Intermolecular Charge Transfer Between Heterocyclic Oligomers. Effects of Heteroatom and Molecular Packing On Hopping Transport in Organic SemiconductorsDfmso0No ratings yet
- Infrared Spectra of Small Insertion and Methylidene Complexes in Reactions of Laser-Ablated Palladium Atoms With HalomethanesDocument9 pagesInfrared Spectra of Small Insertion and Methylidene Complexes in Reactions of Laser-Ablated Palladium Atoms With HalomethanesDiego Alejandro Hurtado BalcazarNo ratings yet
- Study of The Effect Induced by The Substituents On The Ring - Chain Tautomerism of Schiff Bases Derived From NorephedrineDocument7 pagesStudy of The Effect Induced by The Substituents On The Ring - Chain Tautomerism of Schiff Bases Derived From NorephedrineDiogo DiasNo ratings yet
- Alkaline Lyotropic Silicate Surfactant Liquid CrystalsDocument15 pagesAlkaline Lyotropic Silicate Surfactant Liquid Crystalsrestihariyani_160035No ratings yet
- M.sc. - II, Organic ChemistryDocument15 pagesM.sc. - II, Organic ChemistryDeepak50% (2)
- 623 FullDocument5 pages623 Fullmariano totoNo ratings yet
- Inorganic Cluster As Single SourceDocument10 pagesInorganic Cluster As Single Sourceprakush01975225403No ratings yet
- Spatiotemporal Microbial Evolution On Antibiotic Landscapes: 13 ReferencesandnotesDocument6 pagesSpatiotemporal Microbial Evolution On Antibiotic Landscapes: 13 ReferencesandnotesFranco VeglianiNo ratings yet
- Cryocrystallography of Biological Macromolecules - A Generally Applicable MethodDocument5 pagesCryocrystallography of Biological Macromolecules - A Generally Applicable MethodJun Obam'doNo ratings yet
- JM 00129 A 024Document9 pagesJM 00129 A 024Asilentamerican NguyenNo ratings yet
- Cy 5012-2024 Mingos Fusion FormalismDocument9 pagesCy 5012-2024 Mingos Fusion Formalism0066Ritul BhatiaNo ratings yet
- Ando 2005Document4 pagesAndo 2005Lê Đức HuyNo ratings yet
- Assignment CHEM 211Document5 pagesAssignment CHEM 211Leoterio LacapNo ratings yet
- Oso PolarDocument5 pagesOso PolarGiovanni Ávila EsquivelNo ratings yet
- 2,3-Disubstituted Thiophene-Based Organic Dyes For Solar CellsDocument11 pages2,3-Disubstituted Thiophene-Based Organic Dyes For Solar CellsSamita RaiNo ratings yet
- Ligand-Stabilized Ruthenium Nanoparticles: Synthesis, Organization, and DynamicsDocument11 pagesLigand-Stabilized Ruthenium Nanoparticles: Synthesis, Organization, and DynamicsGeorge FuryNo ratings yet
- Ring-Opening Polymerization of Strained, Ring-Tilted Ferrocenophanes: A Route To High Molecular Weight Poly (Ferrocenyki1anes) TDocument3 pagesRing-Opening Polymerization of Strained, Ring-Tilted Ferrocenophanes: A Route To High Molecular Weight Poly (Ferrocenyki1anes) TGuty LaraNo ratings yet
- Isotruxene-Derived Cone-Shaped Organic Dyes For Dye-Sensitized Solar CellsDocument10 pagesIsotruxene-Derived Cone-Shaped Organic Dyes For Dye-Sensitized Solar CellsDiogomussumNo ratings yet
- Organometallics 2005 1173Document11 pagesOrganometallics 2005 1173Filute OptionNo ratings yet
- Morphology of Aggregated Asphaltene Structural ModelsDocument11 pagesMorphology of Aggregated Asphaltene Structural ModelsJonathanNo ratings yet
- Yu Jin Jung Et Al - Dendron Arrays For The Force-Based Detection of DNA Hybridization EventsDocument7 pagesYu Jin Jung Et Al - Dendron Arrays For The Force-Based Detection of DNA Hybridization EventsGlade680No ratings yet
- RetractionDocument4 pagesRetractionmike_brynerNo ratings yet
- Estruturas Do H+Document4 pagesEstruturas Do H+Esteban Lopez MorenoNo ratings yet
- Catalytic Direct Arylation With Aryl Chlorides, Bromides, and Iodides: Intramolecular Studies Leading To New Intermolecular ReactionsDocument10 pagesCatalytic Direct Arylation With Aryl Chlorides, Bromides, and Iodides: Intramolecular Studies Leading To New Intermolecular ReactionsJORGE IVAN CASTRO CASTRONo ratings yet
- P (N (i-Bu) CH CH) N: Nonionic Lewis Base for Promoting the Room-Temperature Synthesis of r,β-Unsaturated Esters, Fluorides, Ketones, and Nitriles Using Wadsworth -Emmons PhosphonatesDocument9 pagesP (N (i-Bu) CH CH) N: Nonionic Lewis Base for Promoting the Room-Temperature Synthesis of r,β-Unsaturated Esters, Fluorides, Ketones, and Nitriles Using Wadsworth -Emmons PhosphonatesDiogomussumNo ratings yet
- A Tethered Niacin-Derived Pincer Complex With A Nickel-Carbon Bond in Lactate RacemaseDocument5 pagesA Tethered Niacin-Derived Pincer Complex With A Nickel-Carbon Bond in Lactate RacemaseBenoît DesguinNo ratings yet
- ReferencesDocument7 pagesReferencesDamien KhooNo ratings yet
- Metal Cluster Catalysis A Kinetic and MeDocument9 pagesMetal Cluster Catalysis A Kinetic and MeGI2015No ratings yet
- Graphene NanoplateletsDocument7 pagesGraphene NanoplateletsAnkita NiranjanNo ratings yet
- Preparation and Physisorption Characterization of - Glucose-Templated Mesoporous Silica Sol-Gel MaterialsDocument7 pagesPreparation and Physisorption Characterization of - Glucose-Templated Mesoporous Silica Sol-Gel MaterialsDwiNo ratings yet
- Concepts & Synthesis: Emphasizing NEW Ideas TO Stimulate Research IN EcologyDocument12 pagesConcepts & Synthesis: Emphasizing NEW Ideas TO Stimulate Research IN Ecologyjuli100% (1)
- M.SC Chemistry PDFDocument32 pagesM.SC Chemistry PDFHassan JavedNo ratings yet
- Kirsten L. Genson Et Al - Interfacial Micellar Structures From Novel Amphiphilic Star PolymersDocument9 pagesKirsten L. Genson Et Al - Interfacial Micellar Structures From Novel Amphiphilic Star PolymersHumdsNo ratings yet
- Lanthanide Amides ( (Me Si) N) Ln (μ-Cl) Li (THF) Catalyzed Hydrophosphonylation of Aryl AldehydesDocument4 pagesLanthanide Amides ( (Me Si) N) Ln (μ-Cl) Li (THF) Catalyzed Hydrophosphonylation of Aryl AldehydesDiogomussumNo ratings yet
- A Concise and Convergent PA-824 PDFDocument4 pagesA Concise and Convergent PA-824 PDFPULIDO PEÑA JOHN SEBASTIANNo ratings yet
- Synthesis of 2-Alkoxy (Aroxy) - 3-Substituted Quinolines by DABCO-Promoted Cyclization of O-Alkynylaryl IsocyanidesDocument3 pagesSynthesis of 2-Alkoxy (Aroxy) - 3-Substituted Quinolines by DABCO-Promoted Cyclization of O-Alkynylaryl IsocyanidesDiogomussumNo ratings yet
- Stereoselective Synthesis of Cyclohexanones Via Phase Transfer Catalyzed Double Addition of Nucleophiles To Divinyl KetonesDocument3 pagesStereoselective Synthesis of Cyclohexanones Via Phase Transfer Catalyzed Double Addition of Nucleophiles To Divinyl KetonesDiogomussumNo ratings yet
- A Strategy To Synthesize Taxol Side Chain and (-) - Epi Cytoxazone Via Chiral Brønsted Acid-Rh (Oac) Co-Catalyzed Enantioselective Three-Component ReactionsDocument4 pagesA Strategy To Synthesize Taxol Side Chain and (-) - Epi Cytoxazone Via Chiral Brønsted Acid-Rh (Oac) Co-Catalyzed Enantioselective Three-Component ReactionsDiogomussumNo ratings yet
- Evans - Tishchenko Coupling of Heteroaryl Aldehydes: J. Org. Chem. 2010, 75, 7475-7478 7475Document4 pagesEvans - Tishchenko Coupling of Heteroaryl Aldehydes: J. Org. Chem. 2010, 75, 7475-7478 7475DiogomussumNo ratings yet
- Efforts Toward Distorted SpiropentanesDocument4 pagesEfforts Toward Distorted SpiropentanesDiogomussumNo ratings yet
- Rh-Catalyzed Oxidative Coupling Between Primary and Secondary Benzamides and Alkynes: Synthesis of Polycyclic AmidesDocument4 pagesRh-Catalyzed Oxidative Coupling Between Primary and Secondary Benzamides and Alkynes: Synthesis of Polycyclic AmidesDiogomussumNo ratings yet
- Stereoselective R, R - Annelation Reactions of 1,3-Dioxan-5-OnesDocument4 pagesStereoselective R, R - Annelation Reactions of 1,3-Dioxan-5-OnesDiogomussumNo ratings yet
- Stereoselective Synthesis of 2,3,6-Trisubstituted Tetrahydropyridines Via TF O-Mediated Grob Fragmentation: Access To Indolizidines (-) - 209I and (-) - 223JDocument3 pagesStereoselective Synthesis of 2,3,6-Trisubstituted Tetrahydropyridines Via TF O-Mediated Grob Fragmentation: Access To Indolizidines (-) - 209I and (-) - 223JDiogomussumNo ratings yet
- Diastereoselective Silver-Catalyzed 1,3-Dipolar Cycloaddition of Azomethine Ylides With Fluorinated ImineDocument4 pagesDiastereoselective Silver-Catalyzed 1,3-Dipolar Cycloaddition of Azomethine Ylides With Fluorinated ImineDiogomussumNo ratings yet
- Cu (Otf) - Mediated Chan-Lam Reaction of Carboxylic Acids To Access Phenolic EstersDocument3 pagesCu (Otf) - Mediated Chan-Lam Reaction of Carboxylic Acids To Access Phenolic EstersDiogomussumNo ratings yet
- A Versatile and Practical Microwave-Assisted Synthesis of Sterically Hindered N-ArylpiperazinesDocument3 pagesA Versatile and Practical Microwave-Assisted Synthesis of Sterically Hindered N-ArylpiperazinesDiogomussumNo ratings yet
- Concise and Enantioselective Total Synthesis of 15-Deoxy-Δ -Prostaglandin JDocument3 pagesConcise and Enantioselective Total Synthesis of 15-Deoxy-Δ -Prostaglandin JDiogomussumNo ratings yet
- Formation of γ-Oxoacids and 1H-Pyrrol-2 (5H) -ones from r,β-Unsaturated Ketones and Ethyl NitroacetateDocument4 pagesFormation of γ-Oxoacids and 1H-Pyrrol-2 (5H) -ones from r,β-Unsaturated Ketones and Ethyl NitroacetateDiogomussumNo ratings yet
- Probing Binding Preferences of DNA and RNA: Backbone Chirality of Thioacetamido-Linked Nucleic Acids and iso-Thioacetamido-Linked Nucleic Acids To Differentiate DNA Versus RNA Selective BindingDocument4 pagesProbing Binding Preferences of DNA and RNA: Backbone Chirality of Thioacetamido-Linked Nucleic Acids and iso-Thioacetamido-Linked Nucleic Acids To Differentiate DNA Versus RNA Selective BindingDiogomussumNo ratings yet
- Castellano2010 PDFDocument4 pagesCastellano2010 PDFGiacomo AccomandoNo ratings yet
- Synthesis of Enantiopure 3-Substituted MorpholinesDocument4 pagesSynthesis of Enantiopure 3-Substituted MorpholinesDiogomussumNo ratings yet
- Synthesis of 2-Alkyl-And Aryl-3-Ethoxycarbonyl-2,5 - Dihydrofurans Through Gold-Catalyzed Intramolecular HydroalkoxylationDocument4 pagesSynthesis of 2-Alkyl-And Aryl-3-Ethoxycarbonyl-2,5 - Dihydrofurans Through Gold-Catalyzed Intramolecular HydroalkoxylationDiogomussumNo ratings yet
- Synthesis of Regioselectively Functionalized Benzo (B) Thiophenes by Combined ortho-Lithiation-Halocyclization StrategiesDocument4 pagesSynthesis of Regioselectively Functionalized Benzo (B) Thiophenes by Combined ortho-Lithiation-Halocyclization StrategiesDiogomussumNo ratings yet
- Facile Synthesis of Koser's Reagent and Derivatives From Iodine or Aryl IodidesDocument4 pagesFacile Synthesis of Koser's Reagent and Derivatives From Iodine or Aryl IodidesDiogomussumNo ratings yet
- Water-Compatible Iminium Activation: Highly Enantioselective Organocatalytic Michael Addition of Malonates to r,β-Unsaturated EnonesDocument3 pagesWater-Compatible Iminium Activation: Highly Enantioselective Organocatalytic Michael Addition of Malonates to r,β-Unsaturated EnonesDiogomussumNo ratings yet
- Regioselective One-Pot Protection of - Glucosamine: 7424 J. Org. Chem. 2010, 75, 7424-7427Document4 pagesRegioselective One-Pot Protection of - Glucosamine: 7424 J. Org. Chem. 2010, 75, 7424-7427DiogomussumNo ratings yet
- Biomimetic Total Synthesis of Meiogynin A, An Inhibitor of BCL-XL and Bak InteractionDocument4 pagesBiomimetic Total Synthesis of Meiogynin A, An Inhibitor of BCL-XL and Bak InteractionDiogomussumNo ratings yet
- Total Synthesis of Clathculins A and BDocument4 pagesTotal Synthesis of Clathculins A and BDiogomussumNo ratings yet
- Synthesis, Structure, and Binding Property of Pentiptycene-Based Rigid Tweezer-Like MoleculesDocument4 pagesSynthesis, Structure, and Binding Property of Pentiptycene-Based Rigid Tweezer-Like MoleculesDiogomussumNo ratings yet
- Access To Resorcylic Acid Lactones Via Phosphonate Based Intramolecular OlefinationDocument4 pagesAccess To Resorcylic Acid Lactones Via Phosphonate Based Intramolecular OlefinationDiogomussumNo ratings yet
- Synthesis of Chlorinated Bicyclic C-Fused Tetrahydrofuro (3,2-c) Azetidin-2-OnesDocument4 pagesSynthesis of Chlorinated Bicyclic C-Fused Tetrahydrofuro (3,2-c) Azetidin-2-OnesDiogomussumNo ratings yet
- Polyfluorination Using IF: Tadahito Fukuhara and Shoji HaraDocument7 pagesPolyfluorination Using IF: Tadahito Fukuhara and Shoji HaraDiogomussumNo ratings yet
- Heteroatom-Directed Reverse Wacker Oxidations. Synthesis of The Reported Structure of (-) - Herbaric AcidDocument5 pagesHeteroatom-Directed Reverse Wacker Oxidations. Synthesis of The Reported Structure of (-) - Herbaric AcidDiogomussumNo ratings yet
- 1491306950894tseamcet 2016 LastranksDocument23 pages1491306950894tseamcet 2016 LastranksMohammed Abdulwajid SiddiquiNo ratings yet
- Centroides PDFDocument7 pagesCentroides PDFDavid FernandoNo ratings yet
- ALFAsim Technical Manual EN USDocument70 pagesALFAsim Technical Manual EN UScleuberNo ratings yet
- Physics Malaysian Matriculation Semester 1 Notes CompleteDocument474 pagesPhysics Malaysian Matriculation Semester 1 Notes CompleteJay Bee96% (68)
- Microwave Engineering-HazardsDocument20 pagesMicrowave Engineering-HazardsKobid KarkeeNo ratings yet
- Exp 2 Graphical Synthesis of Planar MechanismsDocument7 pagesExp 2 Graphical Synthesis of Planar MechanismsPaachi ThakurNo ratings yet
- Physics Division: Phys 141 Syllabus, PGDocument7 pagesPhysics Division: Phys 141 Syllabus, PGJun YoutubeNo ratings yet
- Modular FURNACE INSULATION THKNESS CALDocument2 pagesModular FURNACE INSULATION THKNESS CALRamachandra Bhat HireNo ratings yet
- Study of Electrical Properties of (Pva-Cao) Composites: Ahmed HashimDocument4 pagesStudy of Electrical Properties of (Pva-Cao) Composites: Ahmed HashimMohammad HarirNo ratings yet
- Polymorphic Phase TransitionsDocument24 pagesPolymorphic Phase Transitionsnimmy kumariNo ratings yet
- Task 1 - Esneyder QuevedoDocument11 pagesTask 1 - Esneyder QuevedoEsneyder QuevedoNo ratings yet
- 1 Year Study Plan For IIT JEE MainDocument9 pages1 Year Study Plan For IIT JEE MainShubham kumar100% (1)
- Epr NotesDocument40 pagesEpr NotesArunachalamNo ratings yet
- More Accurate Localized Wire Rope Testing Based On Hall Sensor ArrayDocument2 pagesMore Accurate Localized Wire Rope Testing Based On Hall Sensor ArrayNesanNo ratings yet
- End Plate DesignDocument26 pagesEnd Plate DesignNitesh SinghNo ratings yet
- NEET Physics Quota Neetprep Physics Ques. Physics Ques. 70% Weightage WeightageDocument1 pageNEET Physics Quota Neetprep Physics Ques. Physics Ques. 70% Weightage WeightageNikhilNo ratings yet
- 4.26μm sensor CO2Document9 pages4.26μm sensor CO2JindraNo ratings yet
- Contributions of Indian Physicists in ScienceDocument42 pagesContributions of Indian Physicists in ScienceblahNo ratings yet
- Intruduccion A La FísicaDocument21 pagesIntruduccion A La FísicaRAFAEL TORRESNo ratings yet
- 5 1doneDocument3 pages5 1doneeperlaNo ratings yet
- Nuclear Physics - Mind Map - Lakshya NEET 2024Document1 pageNuclear Physics - Mind Map - Lakshya NEET 2024aanyakaurchonaNo ratings yet
- Essential University Physics 3rd Edition Richard Wolfson Test BankDocument11 pagesEssential University Physics 3rd Edition Richard Wolfson Test Bankfelixedanafte5n100% (32)
- Complex Engineering Problem: Mohammad Hussain Submitted To: BSME: 18-22 Roll: 19 Dr. Rab NawazDocument11 pagesComplex Engineering Problem: Mohammad Hussain Submitted To: BSME: 18-22 Roll: 19 Dr. Rab NawazMohammad HussainNo ratings yet
- Assing 4Document2 pagesAssing 4KazaValiShaikNo ratings yet
- Must Know-Physics (NMAT)Document18 pagesMust Know-Physics (NMAT)iya100% (2)
- Solitons and Chaos I. Antoniou and J. F. LambertDocument340 pagesSolitons and Chaos I. Antoniou and J. F. Lambertmario_gNo ratings yet
- B.N. Lucas, W.C. Oliver, Metall. Mater. Trans. 30ADocument10 pagesB.N. Lucas, W.C. Oliver, Metall. Mater. Trans. 30AFuzeng RenNo ratings yet
- Curva Goulds 3196Document2 pagesCurva Goulds 3196edwinsazzz75% (4)
- Physics - Er. Anil Sir Om Sai Ram Class - 11, 12 & CbseDocument26 pagesPhysics - Er. Anil Sir Om Sai Ram Class - 11, 12 & CbseArnav MishraNo ratings yet
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincFrom EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincRating: 3.5 out of 5 stars3.5/5 (137)
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolFrom EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolNo ratings yet
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeFrom EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeRating: 5 out of 5 stars5/5 (4)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (90)
- Formulating, Packaging, and Marketing of Natural Cosmetic ProductsFrom EverandFormulating, Packaging, and Marketing of Natural Cosmetic ProductsNo ratings yet
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideFrom EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideNo ratings yet
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (5)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeFrom EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeRating: 4 out of 5 stars4/5 (1)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsFrom EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsRating: 4 out of 5 stars4/5 (146)
- AP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeFrom EverandAP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeRating: 5 out of 5 stars5/5 (1)
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeFrom EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeNo ratings yet
- The Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsFrom EverandThe Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsRating: 5 out of 5 stars5/5 (3)
- The Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableFrom EverandThe Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableRating: 3.5 out of 5 stars3.5/5 (22)
- Taste: Surprising Stories and Science About Why Food Tastes GoodFrom EverandTaste: Surprising Stories and Science About Why Food Tastes GoodRating: 3 out of 5 stars3/5 (20)
- The Periodic Table: A Very Short IntroductionFrom EverandThe Periodic Table: A Very Short IntroductionRating: 4.5 out of 5 stars4.5/5 (3)
- Tribology: Friction and Wear of Engineering MaterialsFrom EverandTribology: Friction and Wear of Engineering MaterialsRating: 5 out of 5 stars5/5 (1)
- Water-Based Paint Formulations, Vol. 3From EverandWater-Based Paint Formulations, Vol. 3Rating: 4.5 out of 5 stars4.5/5 (6)
- Ingredients: A Visual Exploration of 75 Additives & 25 Food ProductsFrom EverandIngredients: A Visual Exploration of 75 Additives & 25 Food ProductsRating: 4 out of 5 stars4/5 (1)
- The Periodic Table of Elements - Alkali Metals, Alkaline Earth Metals and Transition Metals | Children's Chemistry BookFrom EverandThe Periodic Table of Elements - Alkali Metals, Alkaline Earth Metals and Transition Metals | Children's Chemistry BookNo ratings yet
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (14)
- Bioplastics: A Home Inventors HandbookFrom EverandBioplastics: A Home Inventors HandbookRating: 4 out of 5 stars4/5 (2)
- High School Chemistry: Comprehensive Content for High School ChemistryFrom EverandHigh School Chemistry: Comprehensive Content for High School ChemistryNo ratings yet
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (1)
- Chemistry: a QuickStudy Laminated Reference GuideFrom EverandChemistry: a QuickStudy Laminated Reference GuideRating: 5 out of 5 stars5/5 (1)