You might also like
- Chronic Lymphocytic LeukemiaFrom EverandChronic Lymphocytic LeukemiaMichael HallekNo ratings yet
- STAT-6 in Hodgkin Lymphoma Pathobiology and Treatment-Review of The LiteratureDocument8 pagesSTAT-6 in Hodgkin Lymphoma Pathobiology and Treatment-Review of The LiteratureEditor SaraNo ratings yet
- Bao 2Document12 pagesBao 2Phạm ĐứcNo ratings yet
- Linfoma de HodgkinDocument25 pagesLinfoma de HodgkinPierrotNo ratings yet
- Dieta y LeucemiaDocument53 pagesDieta y LeucemiaJose Angel AbadíaNo ratings yet
- Chronic Lymphocytic Leukemia: A Clinical and Molecular Heterogenous DiseaseDocument14 pagesChronic Lymphocytic Leukemia: A Clinical and Molecular Heterogenous DiseaseCallisthenisLeventisNo ratings yet
- Reviews: The Biology of Hodgkin's LymphomaDocument13 pagesReviews: The Biology of Hodgkin's LymphomaPhạm ĐứcNo ratings yet
- Recent Advances in Langerhans Cell HistiDocument11 pagesRecent Advances in Langerhans Cell HistiThanasis PapatheodorouNo ratings yet
- Jurnal Patofisiologi Limfoma HodkinDocument8 pagesJurnal Patofisiologi Limfoma HodkinRedy RohmansyahNo ratings yet
- Lab 1 (Article)Document5 pagesLab 1 (Article)Vignesh R.PuvanesuaranNo ratings yet
- Chronic Lymphocytic Leukemia (CLL) - Then and Now: Critical ReviewDocument11 pagesChronic Lymphocytic Leukemia (CLL) - Then and Now: Critical ReviewJose Angel AbadíaNo ratings yet
- AbdfDocument9 pagesAbdfXONNo ratings yet
- Non Hodgkin LymphomaDocument9 pagesNon Hodgkin LymphomaAhmad SaifulNo ratings yet
- Transformation of Follicular Lymphoma To Hodgkin-Like Lymphoma: A Case Report of A Diagnostic PitfallDocument4 pagesTransformation of Follicular Lymphoma To Hodgkin-Like Lymphoma: A Case Report of A Diagnostic PitfallIJAR JOURNALNo ratings yet
- Diagnosis of Hodgkins Disease An Update On Histopathological and Immunophenotypical FeaturesDocument13 pagesDiagnosis of Hodgkins Disease An Update On Histopathological and Immunophenotypical Featuresyv55xfwrpcNo ratings yet
- Bmi s16553Document8 pagesBmi s16553Cassandra VérasNo ratings yet
- Non-Hodgkin's Lymphoma: A Histopathologic and Prognostic EvaluationDocument12 pagesNon-Hodgkin's Lymphoma: A Histopathologic and Prognostic Evaluationchimbimb100% (2)
- J Immunol-2003-Woo-6273-9Document8 pagesJ Immunol-2003-Woo-6273-9MuhammadGagasSasongkoNo ratings yet
- Langerhans Cell HistiocytosisDocument7 pagesLangerhans Cell HistiocytosisDarwin ElguiraNo ratings yet
- Malignant LymphomaDocument24 pagesMalignant LymphomaalifiaNo ratings yet
- IHC LymphomaDocument21 pagesIHC LymphomaNGUYEN QUYNHNo ratings yet
- ArtLinfoma InfolenteDocument5 pagesArtLinfoma InfolenteSamy De La Cruz SánchezNo ratings yet
- Articol MYD88, PIM-1Document18 pagesArticol MYD88, PIM-1Ionut PoinareanuNo ratings yet
- Final Review Med.J of Hem and Inf DiseasesDocument14 pagesFinal Review Med.J of Hem and Inf Diseasesmgounari7293No ratings yet
- HLA Sensitisation - Can It Be PreventedDocument11 pagesHLA Sensitisation - Can It Be PreventedPatriciaNo ratings yet
- 1 s2.0 S0145212612004456 MainDocument9 pages1 s2.0 S0145212612004456 MainMilana PerkovićNo ratings yet
- MDSC en Enfermedades HematologicasDocument12 pagesMDSC en Enfermedades HematologicasLyanna StarkNo ratings yet
- Aberrations of MYC Are A Common Event in B-Cell Prolymphocytic LeukemiaDocument9 pagesAberrations of MYC Are A Common Event in B-Cell Prolymphocytic LeukemiaRafa AssidiqNo ratings yet
- Rodrguez Vicente 2013Document14 pagesRodrguez Vicente 2013Jose AbadiaNo ratings yet
- Impact and Intricacies of Bone Marrow Microenvironment in B-Cell Lymphomas: From Biology To TherapyDocument19 pagesImpact and Intricacies of Bone Marrow Microenvironment in B-Cell Lymphomas: From Biology To TherapysayansoccercrazyNo ratings yet
- Langerhans Cell Histiocytosis Followed by Folliculotropic Mycosis FungoidesDocument4 pagesLangerhans Cell Histiocytosis Followed by Folliculotropic Mycosis FungoidesYuniita VerayantiiNo ratings yet
- Tse 2017Document13 pagesTse 2017Ke XuNo ratings yet
- Reviews: Chronic Lymphocytic Leukaemia: From Genetics To TreatmentDocument18 pagesReviews: Chronic Lymphocytic Leukaemia: From Genetics To Treatmentlengers poworNo ratings yet
- Bosch2019 PDFDocument18 pagesBosch2019 PDFlengers poworNo ratings yet
- HLH ReviewDocument13 pagesHLH ReviewGabriel Arturo Tucto CastañedaNo ratings yet
- Artigo 8Document9 pagesArtigo 8raudneimNo ratings yet
- Atypical Cellular Disorders: Kenneth L. Mcclain, Yasodha Natkunam, and Steven H. SwerdlowDocument14 pagesAtypical Cellular Disorders: Kenneth L. Mcclain, Yasodha Natkunam, and Steven H. SwerdlowhemendreNo ratings yet
- Cancers 11 00Document17 pagesCancers 11 00GeorgeNo ratings yet
- Hodgkin LyphomaDocument6 pagesHodgkin LyphomaJose Luis PittauNo ratings yet
- Diffuse Large B-Cell Lymphoma: The History, Current View and New PerspectivesDocument14 pagesDiffuse Large B-Cell Lymphoma: The History, Current View and New PerspectivesPepe PintoNo ratings yet
- 10 5772@54652 PDFDocument32 pages10 5772@54652 PDFhollNo ratings yet
- Novel Targeted Therapies For Chronic Lymphocytic Leukemia in Elderly Patients: A Systematic ReviewDocument13 pagesNovel Targeted Therapies For Chronic Lymphocytic Leukemia in Elderly Patients: A Systematic ReviewVishal BondeNo ratings yet
- LinfomaDocument9 pagesLinfomaRocio HaroNo ratings yet
- Bone Marrow Manifestation of Plasmablastic Transformation of Chronic Lymphocytic Leukemia: A Case ReportDocument5 pagesBone Marrow Manifestation of Plasmablastic Transformation of Chronic Lymphocytic Leukemia: A Case ReportTomasz GórskiNo ratings yet
- After CorrectionDocument22 pagesAfter CorrectionRania GebbaNo ratings yet
- Aggressive Lymphomas NEJMDocument13 pagesAggressive Lymphomas NEJMAnnaNo ratings yet
- Pathogenesis and Prognostication in Acute Lymphoblastic LeukemiaDocument6 pagesPathogenesis and Prognostication in Acute Lymphoblastic Leukemiamarvin lionelNo ratings yet
- Encyclopedia 02 00061Document9 pagesEncyclopedia 02 00061Ritwik DasNo ratings yet
- Lymphoma HerfindalDocument56 pagesLymphoma HerfindalAanshi ShahNo ratings yet
- Successful Management of Granulocytic Sarcoma With Co - 2016 - Pediatric HematolDocument2 pagesSuccessful Management of Granulocytic Sarcoma With Co - 2016 - Pediatric HematolHawin NurdianaNo ratings yet
- Overview of LymphomaDocument42 pagesOverview of Lymphomaadamu mohammadNo ratings yet
- Pasq 2004 BloodDocument8 pagesPasq 2004 BloodlillareinigerNo ratings yet
- The Role of Cytokines in Acute Myeloid Leukemia: A Systematic ReviewDocument12 pagesThe Role of Cytokines in Acute Myeloid Leukemia: A Systematic Reviewhoangphuong08101992No ratings yet
- Mutaz Et Al Cancers 2022Document21 pagesMutaz Et Al Cancers 2022Farid JohanNo ratings yet
- PathogenesisDocument14 pagesPathogenesisIdreesNo ratings yet
- Molecular Evidence For Antigen-Driven Immune Responses in Cardiac Lesions of Rheumatic Heart Disease PatientsDocument12 pagesMolecular Evidence For Antigen-Driven Immune Responses in Cardiac Lesions of Rheumatic Heart Disease PatientsJonasNo ratings yet
- Immunology in Transplantation - Basics For BeginnersDocument6 pagesImmunology in Transplantation - Basics For BeginnersTuan Thanh NguyenNo ratings yet
- Hodgkin Lymphoma 1Document17 pagesHodgkin Lymphoma 1taufik perdanaNo ratings yet
- 1999 DLBCL CD56Document7 pages1999 DLBCL CD56maomaochongNo ratings yet
- 2016-Yahav Et Al-Cytometry Part ADocument10 pages2016-Yahav Et Al-Cytometry Part AgiladNo ratings yet
- Abbott 2017Document5 pagesAbbott 2017Paola Alexandra Chavez FNo ratings yet
- Anemia Aplasica - BMJDocument21 pagesAnemia Aplasica - BMJPaola Alexandra Chavez FNo ratings yet
- International Journal of Surgery Case ReportsDocument4 pagesInternational Journal of Surgery Case ReportsPaola Alexandra Chavez FNo ratings yet
- ITU Niños Lineamientos Europeos. EU 2014Document13 pagesITU Niños Lineamientos Europeos. EU 2014KathyNo ratings yet
- Nasal Bone Fractures: DR V.Ramkumar Consultant Dental&Faciomaxillary Surgeon Reg No: 4118-Tamilnadu-India (Asia)Document10 pagesNasal Bone Fractures: DR V.Ramkumar Consultant Dental&Faciomaxillary Surgeon Reg No: 4118-Tamilnadu-India (Asia)Paola Alexandra Chavez FNo ratings yet
- 2012 - Erectile Dysfunction in Arab CountriesDocument7 pages2012 - Erectile Dysfunction in Arab CountriesPaola Alexandra Chavez FNo ratings yet
- Tuberculosis NEJMDocument25 pagesTuberculosis NEJMPaola Alexandra Chavez F100% (1)
- Mycobacterium TuberculosisDocument34 pagesMycobacterium TuberculosisPaola Alexandra Chavez FNo ratings yet
- Tuberculosis PathogenesisDocument26 pagesTuberculosis PathogenesisAfrisya Bimo SiwendroNo ratings yet
- TuberculosisDocument55 pagesTuberculosisPaola Alexandra Chavez FNo ratings yet
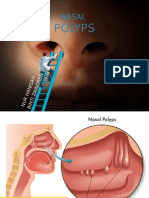
- Nasal PolypsDocument27 pagesNasal PolypsPaola Alexandra Chavez F50% (2)
- STRA65Document6 pagesSTRA65SUJITH232323No ratings yet
- Maria Immaculata Iwo: Sekolah Farmasi ItbDocument55 pagesMaria Immaculata Iwo: Sekolah Farmasi ItbEkwan Prasetyo AzlinNo ratings yet
- Thiazolopyrimidines: A Retrospective Study of Synthesis, Structure-Activity Relationship and Diverse Pharmacological ActionsDocument12 pagesThiazolopyrimidines: A Retrospective Study of Synthesis, Structure-Activity Relationship and Diverse Pharmacological ActionsRajat GoyalNo ratings yet
- Pratiwi S Gunawan, BDS., DDS., MDSC, PHDDocument53 pagesPratiwi S Gunawan, BDS., DDS., MDSC, PHDBima Ewando KabanNo ratings yet
- Approved StemsDocument2 pagesApproved StemsRajesh singh BhatiNo ratings yet
- Cerri Et Al. 2017Document7 pagesCerri Et Al. 2017JD SánchezNo ratings yet
- Art:10.1186/1939 4551 1 S3 A1 PDFDocument335 pagesArt:10.1186/1939 4551 1 S3 A1 PDFYunus wanendaNo ratings yet
- Dermatologic Immunity PDFDocument391 pagesDermatologic Immunity PDFChino UnjbgNo ratings yet
- McGraw-Hill Education NAPLEX Review GuideDocument77 pagesMcGraw-Hill Education NAPLEX Review Guidebobfoo100% (1)
- Crossword ImmunoDocument1 pageCrossword Immunoerasmoz9222No ratings yet
- Clinical Flow Cytometry - Emerging ApplicationsDocument215 pagesClinical Flow Cytometry - Emerging ApplicationsDaoud IssaNo ratings yet
- Association Between Polymorphisms in CXCR2 Gene and PreeclampsiaDocument8 pagesAssociation Between Polymorphisms in CXCR2 Gene and PreeclampsiaalyneNo ratings yet
- Rheumatic Diseases Immunological Mechanisms and Prospects For New TherapiesDocument297 pagesRheumatic Diseases Immunological Mechanisms and Prospects For New TherapiesАндрей ТихомировNo ratings yet
- Fistula ArteriovenosaDocument14 pagesFistula ArteriovenosaJoshua CajinaNo ratings yet
- Techniques For Immune Function Analysis HandbookDocument248 pagesTechniques For Immune Function Analysis HandbookDante AvilésNo ratings yet
- Tissue Eng ToolsDocument20 pagesTissue Eng ToolsVijay RajNo ratings yet
- Design, Synthesis, and Biological Evaluation of The Combinatorial Library With A NewDocument13 pagesDesign, Synthesis, and Biological Evaluation of The Combinatorial Library With A NewSimon LeluyerNo ratings yet
- Kuby Immunology 7e TBDocument134 pagesKuby Immunology 7e TBStarrx71494% (18)
- Stine Helene Falsig Pedersen (Editor) - Reviews of Physiology, Biochemistry and Pharmacology. 177-Springer (2021)Document155 pagesStine Helene Falsig Pedersen (Editor) - Reviews of Physiology, Biochemistry and Pharmacology. 177-Springer (2021)shuvvro dhaNo ratings yet
- Acute Inflammatory Response - : VPM 152 Winter 2006 Inflammation and Repair General Pathology 18Document19 pagesAcute Inflammatory Response - : VPM 152 Winter 2006 Inflammation and Repair General Pathology 18ANI REHMNo ratings yet
- 1 s2.0 S0085253822010171 MainDocument15 pages1 s2.0 S0085253822010171 MainJack JackNo ratings yet
- Human Body Temperature and New Approaches To ConstDocument21 pagesHuman Body Temperature and New Approaches To ConstCarlos Apodman BellaNo ratings yet
- Current Status of Novel Antifibrotic Therapies in Patients With Chronic Liver DiseaseDocument27 pagesCurrent Status of Novel Antifibrotic Therapies in Patients With Chronic Liver DiseaseGhenea Catalin-StefanNo ratings yet
- An Overview of The Immune SystemDocument14 pagesAn Overview of The Immune SystemMaría Camila ValenciaNo ratings yet
- Inmunologia Repaso Capitulo 9 UprrpDocument23 pagesInmunologia Repaso Capitulo 9 UprrpAlexandra Leonor Pujols AstacioNo ratings yet
- Kocluhetemoglu 2018Document5 pagesKocluhetemoglu 2018adNo ratings yet
- Acute & Chronic InflammationDocument102 pagesAcute & Chronic InflammationVenz100% (1)
- Robbins Pathology - Chapter 3 TransDocument18 pagesRobbins Pathology - Chapter 3 Transnath nath100% (7)
- Kornman, Page, Tonetti 1997-Periodontology - 2000Document22 pagesKornman, Page, Tonetti 1997-Periodontology - 2000Liliana Miranda ANo ratings yet
- Inflammatory Mediators in The Pathogenesis of Periodontitis: Expert ReviewsDocument22 pagesInflammatory Mediators in The Pathogenesis of Periodontitis: Expert ReviewsPriya MalhotraNo ratings yet
- The Age of Magical Overthinking: Notes on Modern IrrationalityFrom EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityRating: 4 out of 5 stars4/5 (24)
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisFrom EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisRating: 4.5 out of 5 stars4.5/5 (42)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsFrom EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsNo ratings yet
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedFrom EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedRating: 5 out of 5 stars5/5 (80)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsFrom EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsRating: 5 out of 5 stars5/5 (1)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaFrom EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaRating: 4.5 out of 5 stars4.5/5 (266)
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeFrom EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeRating: 2 out of 5 stars2/5 (1)
- The Obesity Code: Unlocking the Secrets of Weight LossFrom EverandThe Obesity Code: Unlocking the Secrets of Weight LossRating: 4 out of 5 stars4/5 (5)
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4 out of 5 stars4/5 (3)
- Sleep Stories for Adults: Overcome Insomnia and Find a Peaceful AwakeningFrom EverandSleep Stories for Adults: Overcome Insomnia and Find a Peaceful AwakeningRating: 4 out of 5 stars4/5 (3)
- Gut: the new and revised Sunday Times bestsellerFrom EverandGut: the new and revised Sunday Times bestsellerRating: 4 out of 5 stars4/5 (392)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.From EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Rating: 4.5 out of 5 stars4.5/5 (110)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsFrom EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsRating: 3.5 out of 5 stars3.5/5 (3)
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDFrom EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDRating: 5 out of 5 stars5/5 (1)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisFrom EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisRating: 3.5 out of 5 stars3.5/5 (2)
- An Autobiography of Trauma: A Healing JourneyFrom EverandAn Autobiography of Trauma: A Healing JourneyRating: 5 out of 5 stars5/5 (2)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsFrom EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsRating: 4.5 out of 5 stars4.5/5 (169)
- Mindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessFrom EverandMindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessRating: 4.5 out of 5 stars4.5/5 (328)
- Outlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisFrom EverandOutlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisRating: 4 out of 5 stars4/5 (1)
- Summary: The Myth of Normal: Trauma, Illness, and Healing in a Toxic Culture By Gabor Maté MD & Daniel Maté: Key Takeaways, Summary & AnalysisFrom EverandSummary: The Myth of Normal: Trauma, Illness, and Healing in a Toxic Culture By Gabor Maté MD & Daniel Maté: Key Takeaways, Summary & AnalysisRating: 4 out of 5 stars4/5 (9)
- 12 Rules for Life by Jordan B. Peterson - Book Summary: An Antidote to ChaosFrom Everand12 Rules for Life by Jordan B. Peterson - Book Summary: An Antidote to ChaosRating: 4.5 out of 5 stars4.5/5 (207)
- Cult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryFrom EverandCult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryRating: 4 out of 5 stars4/5 (44)
- The Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeFrom EverandThe Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeRating: 4.5 out of 5 stars4.5/5 (253)