You might also like
- Mécanique Analytique Cours en Français 2020-2021Document105 pagesMécanique Analytique Cours en Français 2020-2021Marius BrunetNo ratings yet
- Proprietés Physique Meca Des MateriauxDocument206 pagesProprietés Physique Meca Des Materiauxabderazak_2008100% (1)
- StereochimieDocument17 pagesStereochimieDarel NadjieraNo ratings yet
- SoufreDocument8 pagesSoufreDarel NadjieraNo ratings yet
- CCMDocument4 pagesCCMDarel NadjieraNo ratings yet
- MetauxDocument13 pagesMetauxDarel NadjieraNo ratings yet
- Derives CarbonylesDocument31 pagesDerives CarbonylesDarel Nadjiera100% (1)
- ChloreDocument18 pagesChloreDarel NadjieraNo ratings yet
- CCMDocument4 pagesCCMDarel NadjieraNo ratings yet
- ChloreDocument18 pagesChloreDarel NadjieraNo ratings yet
- RpeDocument47 pagesRpeDarel Nadjiera100% (1)
- Échange D'ions - Principes de BaseDocument22 pagesÉchange D'ions - Principes de BaseKFJDONo ratings yet
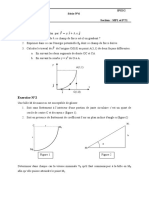
- Révisions Physique 3D - Exercices 31-05-20Document30 pagesRévisions Physique 3D - Exercices 31-05-20Mentalité ArchaïqueNo ratings yet
- Partie I ElectromagnétiqueDocument31 pagesPartie I Electromagnétiquealeatoire buzzNo ratings yet
- TD Magnétostatique-Série 4-2021Document5 pagesTD Magnétostatique-Série 4-2021Zakaria MJNo ratings yet
- Togo 2016 Phyiques Series CEDocument4 pagesTogo 2016 Phyiques Series CEHAMADA1972100% (1)
- SEQ 5 - Stabilité Des Entités ChimiquesDocument4 pagesSEQ 5 - Stabilité Des Entités ChimiquesSpectre UnNo ratings yet
- Les Mesures en ChimieDocument6 pagesLes Mesures en ChimieamalNo ratings yet
- Travaux Pratique SMC3 Chimie Descriptiv1Document3 pagesTravaux Pratique SMC3 Chimie Descriptiv1Yassine RakchoNo ratings yet
- 5 Déversement CommentairesDocument105 pages5 Déversement CommentairesRussel Kamwa SohNo ratings yet
- Travail Pratique Sur L'Acidification Et La FermentationDocument8 pagesTravail Pratique Sur L'Acidification Et La FermentationmushiziNo ratings yet
- DS1 Chap1Document1 pageDS1 Chap1valdxNo ratings yet
- THT-63-2T-12.5-14º-F-400 IE3: Courbe Caractéristique Et Acoustique Pour 1,2Kg/MDocument3 pagesTHT-63-2T-12.5-14º-F-400 IE3: Courbe Caractéristique Et Acoustique Pour 1,2Kg/MMohamed MouslimNo ratings yet
- Crushing FinalDocument57 pagesCrushing Finalsheritier80No ratings yet
- Qualification de Revetements de SurfaceDocument4 pagesQualification de Revetements de SurfaceZANZANNo ratings yet
- Cours de Thermodynamique Appliquée - TS STGI 1-1Document74 pagesCours de Thermodynamique Appliquée - TS STGI 1-1francky scottNo ratings yet
- Actions Sur ParoiDocument9 pagesActions Sur ParoiMed ElouartiNo ratings yet
- Fiche Transport Addition de Poids ExerciceDocument11 pagesFiche Transport Addition de Poids ExercicezfodhilNo ratings yet
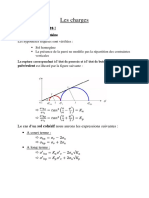
- Résumé Chapitre 1 - Force Et Charge ÉlectriquesDocument3 pagesRésumé Chapitre 1 - Force Et Charge ÉlectriquesHicham noble100% (1)
- Bonneau-Jeanmotte-Leon TDC Rapport Edk CompressedDocument59 pagesBonneau-Jeanmotte-Leon TDC Rapport Edk Compressedapi-638155363No ratings yet
- Amines AliphatiquesDocument7 pagesAmines AliphatiquesOussam OuadidiNo ratings yet
- Referentiels Non Galileens ExercicesDocument6 pagesReferentiels Non Galileens ExercicesRobio GaminGNo ratings yet
- ThermodynamiqueDocument65 pagesThermodynamiqueulrich victor kuateNo ratings yet
- 08 Evolution SpontaneeDocument3 pages08 Evolution SpontaneeChartier JulienNo ratings yet
- Série Théorèmes GénérauxDocument3 pagesSérie Théorèmes Générauxsalem nourNo ratings yet
- Série D'exercices ElectrostatDocument2 pagesSérie D'exercices ElectrostatKheria Zitouni100% (3)
- E211 - Electrocinéttique 1 DipôlesDocument23 pagesE211 - Electrocinéttique 1 DipôlesM'hammed AbouzianeNo ratings yet
- SP00700 DG 1 Water AnalysisDocument4 pagesSP00700 DG 1 Water AnalysisMick VNo ratings yet
- Conditionnement AirDocument10 pagesConditionnement AirWalid Ch-fNo ratings yet
- Chapitre1 Notions de Base de L'électricité - TirageDocument7 pagesChapitre1 Notions de Base de L'électricité - TiragenoussanasnoussaNo ratings yet