You might also like
- Oligonucleotide-Based Drugs and Therapeutics: Preclinical and Clinical Considerations for DevelopmentFrom EverandOligonucleotide-Based Drugs and Therapeutics: Preclinical and Clinical Considerations for DevelopmentNicolay FerrariNo ratings yet
- DNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensFrom EverandDNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensNo ratings yet
- Principle of The PCR: This Technique Is Invented in 1984 by Kary Mulis. The Purpose of A PCRDocument11 pagesPrinciple of The PCR: This Technique Is Invented in 1984 by Kary Mulis. The Purpose of A PCRantesar reheemNo ratings yet
- Protocol ATAC SeqDocument8 pagesProtocol ATAC Seqcnoutsos316No ratings yet
- Polymerase Chain ReactionDocument8 pagesPolymerase Chain ReactionAdigun TaofeeqatNo ratings yet
- Early History of Microbiological TechniqueDocument5 pagesEarly History of Microbiological TechniqueFadare Shadrach ONo ratings yet
- Unit 1 Amplification TechniqueDocument9 pagesUnit 1 Amplification TechniqueShin BoyceNo ratings yet
- Quantifying RNA With QRT-PCR: 2-3 HoursDocument12 pagesQuantifying RNA With QRT-PCR: 2-3 Hourszigurat00No ratings yet
- Polymerase Chain Reaction PCR - Final Report 2 - AlaaDocument9 pagesPolymerase Chain Reaction PCR - Final Report 2 - AlaaAlaa SaadNo ratings yet
- BFP To GFPDocument11 pagesBFP To GFPKeri Gobin SamarooNo ratings yet
- Primer Designing For PCRDocument13 pagesPrimer Designing For PCRiftikharsubha5No ratings yet
- Polymerase Chain Reaction (PCR) - Principle, Steps, ApplicationsDocument5 pagesPolymerase Chain Reaction (PCR) - Principle, Steps, ApplicationsMokhtarCheikhNo ratings yet
- A Basic Polymerase Chain Reaction Protocol: BioinformaticsDocument4 pagesA Basic Polymerase Chain Reaction Protocol: BioinformaticsManish MalikNo ratings yet
- Polymerase Chain Reaction: Determining The Right Magnesium Chloride Concentration For DNA Amplification of Acetobacter XylinumDocument5 pagesPolymerase Chain Reaction: Determining The Right Magnesium Chloride Concentration For DNA Amplification of Acetobacter XylinumAlyza Joy RamirezNo ratings yet
- Definitive PCRDocument55 pagesDefinitive PCRSumit MitraNo ratings yet
- PCR Labwork 2 ENGDocument4 pagesPCR Labwork 2 ENGmigas1996No ratings yet
- DNA Methylation Analysis by Bisulfite Conversion, Cloning, and Sequencing of Individual ClonesDocument11 pagesDNA Methylation Analysis by Bisulfite Conversion, Cloning, and Sequencing of Individual ClonesMacarena Tapia MaldonadoNo ratings yet
- PCR PresentationDocument34 pagesPCR Presentationdurgaprasadhembram1999No ratings yet
- Genomewalker Universal Kit User Manual: Cat. No. 638904 Pt3042-1 (Pr742239) Published 25 April 2007Document30 pagesGenomewalker Universal Kit User Manual: Cat. No. 638904 Pt3042-1 (Pr742239) Published 25 April 2007xprakashNo ratings yet
- Polymerase Chain ReactionDocument34 pagesPolymerase Chain Reactionmokshgoyal2597100% (4)
- Basic PCR ProtocolDocument4 pagesBasic PCR ProtocolEmma MedinaNo ratings yet
- Polymerase Chain Reaction 1Document8 pagesPolymerase Chain Reaction 1Ye Htut AungNo ratings yet
- DNA RecombinationnewDocument34 pagesDNA Recombinationnewsecretary.kimeniahNo ratings yet
- Probe (TTAGGG) N Generated By: Improved Telomere Detection Using Telomere PCRDocument1 pageProbe (TTAGGG) N Generated By: Improved Telomere Detection Using Telomere PCRalejandroNo ratings yet
- NASBADocument16 pagesNASBALalan HolalaNo ratings yet
- DNA Amplification and PCR: by Sara Haydar Mohammed Hasan Fourth Stage A2 Morning StudyDocument9 pagesDNA Amplification and PCR: by Sara Haydar Mohammed Hasan Fourth Stage A2 Morning Studydaloo dmdmNo ratings yet
- Of General Direct Cloning of POR: Construction T-Vectors, A Rapid and System For Unmodified ProductsDocument1 pageOf General Direct Cloning of POR: Construction T-Vectors, A Rapid and System For Unmodified ProductsalejandroNo ratings yet
- Reverse Transcription of The Ribonucleic AcidDocument2 pagesReverse Transcription of The Ribonucleic AcidOmeyya TanveerNo ratings yet
- Polymerase Chain Reaction 243Document1 pagePolymerase Chain Reaction 243Garcia RaphNo ratings yet
- Solanum Lycopersicum Cryptochrome 1 Expression in Response To Short and Long PhotoperiodsDocument22 pagesSolanum Lycopersicum Cryptochrome 1 Expression in Response To Short and Long PhotoperiodsPatrick StumpsNo ratings yet
- PCRDocument5 pagesPCROh RoopzNo ratings yet
- DNA Detection Using Recombination Proteins: BiologyDocument7 pagesDNA Detection Using Recombination Proteins: BiologyclchioNo ratings yet
- Corresponding Author, Email: Matz@whitney - Ufl.eduDocument12 pagesCorresponding Author, Email: Matz@whitney - Ufl.eduDiksha GahlotNo ratings yet
- New Tool For Metabolic Pathway Engineering in Escherichia Coli: One-Step Method To Modulate Expression of Chromosomal GenesDocument5 pagesNew Tool For Metabolic Pathway Engineering in Escherichia Coli: One-Step Method To Modulate Expression of Chromosomal Genesaib_himaniNo ratings yet
- Biotechnology: A. S. M. GiasuddinDocument4 pagesBiotechnology: A. S. M. GiasuddinDespoina ChatziNo ratings yet
- Group 4: DIGITAL PCRDocument14 pagesGroup 4: DIGITAL PCRMaria amor MacapallagNo ratings yet
- Case Report DiagmolDocument12 pagesCase Report DiagmolDina Rizky Amalia ArifNo ratings yet
- PCR and Primer Design CorrectedDocument23 pagesPCR and Primer Design CorrectedAkinade AdetolaNo ratings yet
- Nucleic Acid Amplification PDFDocument9 pagesNucleic Acid Amplification PDFSajeebChandraNo ratings yet
- Amrizal Shalahudin - 20106040026 - Tahapan-Tahapan Metode Site-Directed Mutagenesis - Id.en PDFDocument3 pagesAmrizal Shalahudin - 20106040026 - Tahapan-Tahapan Metode Site-Directed Mutagenesis - Id.en PDFWildan AlfianNo ratings yet
- High-Throughput Droplet Digital PCR System For Absolute PDFDocument7 pagesHigh-Throughput Droplet Digital PCR System For Absolute PDFLuong Nguyen100% (1)
- Chromatin Immunoprecipitation (Chip) : Michael F. Carey, Craig L. Peterson and Stephen T. SmaleDocument9 pagesChromatin Immunoprecipitation (Chip) : Michael F. Carey, Craig L. Peterson and Stephen T. SmaleSwati2013No ratings yet
- Analiza PCR - ThermocyclerDocument7 pagesAnaliza PCR - Thermocyclerionescu_ion_3No ratings yet
- PCR PDFDocument5 pagesPCR PDFrejin rejinrNo ratings yet
- Bauer Core Real Time GuidelinesDocument5 pagesBauer Core Real Time GuidelinesISmi MumtaZahNo ratings yet
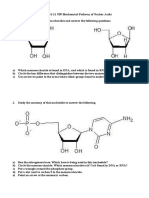
- SEHH2231-22 T09 Biochemical Pathway of Nucleic Acid Exercise (Question)Document6 pagesSEHH2231-22 T09 Biochemical Pathway of Nucleic Acid Exercise (Question)Brian Chi Yan Cheng 鄭智仁No ratings yet
- Directed Evolution of Protein-Based Neurotransmitter Sensors For MRIDocument13 pagesDirected Evolution of Protein-Based Neurotransmitter Sensors For MRIAditya RaghunandanNo ratings yet
- Mathematical Considerations of Competitive Reaction Polymerase ChainDocument11 pagesMathematical Considerations of Competitive Reaction Polymerase ChainNatália RochaNo ratings yet
- Molecular Pathology Techniques: Clinics in Laboratory Medicine December 2013Document21 pagesMolecular Pathology Techniques: Clinics in Laboratory Medicine December 2013Nguyễn HuyềnNo ratings yet
- Southwestern BlottingDocument3 pagesSouthwestern Blottingheran_1006No ratings yet
- BIO 415 DNA Sequencing Part 1 WEEK 12 14 DecDocument27 pagesBIO 415 DNA Sequencing Part 1 WEEK 12 14 DecLejla MillerNo ratings yet
- Biochem Lec 3Document97 pagesBiochem Lec 3yashika gargNo ratings yet
- TMP F936Document3 pagesTMP F936FrontiersNo ratings yet
- 5.2.15-Abt 605 Molecular Diagnostics-Term Paper-PyrosequencingDocument30 pages5.2.15-Abt 605 Molecular Diagnostics-Term Paper-PyrosequencingVinod SivadasanNo ratings yet
- Analytical Biochemistry 1990 Engelke Taq PurificationDocument5 pagesAnalytical Biochemistry 1990 Engelke Taq PurificationPablo L. Cossio RodriguezNo ratings yet
- Polymerase Chain Reaction (PCR) - Principle, Steps, ApplicationsDocument10 pagesPolymerase Chain Reaction (PCR) - Principle, Steps, ApplicationsDr Praveen PrashantNo ratings yet
- Journal of Microbiological Methods 65 (2006) 258 - 267Document10 pagesJournal of Microbiological Methods 65 (2006) 258 - 267dave_owNo ratings yet
- Methods and Instruments For Continuous-Flow PCR On A Chip: Fig. 1: Diagram of The PCR ProcessDocument7 pagesMethods and Instruments For Continuous-Flow PCR On A Chip: Fig. 1: Diagram of The PCR ProcessnanateNo ratings yet
- Deteksi Molekuler 2Document10 pagesDeteksi Molekuler 2CHAYRA STUDIONo ratings yet
- Bleidorn 2017Document18 pagesBleidorn 2017Felipe CardosoNo ratings yet
- Life Sciences and Medicine - Round 1 - Editing TestDocument5 pagesLife Sciences and Medicine - Round 1 - Editing TestUr's Buddy0% (2)
- Study Strategies For BMB 401 or How To Get A Good Grade in Biochemistry?Document6 pagesStudy Strategies For BMB 401 or How To Get A Good Grade in Biochemistry?E12H12No ratings yet
- Alpaca Articles by Alpaca World MagazineDocument6 pagesAlpaca Articles by Alpaca World MagazineDarwin Antezana De la RosaNo ratings yet
- CauveryMK - CVDocument3 pagesCauveryMK - CVMichelle KnightNo ratings yet
- University of Cambridge International Examinations International General Certificate of Secondary Education Biology Paper 1 Multiple Choice October/November 2004 45 MinutesDocument16 pagesUniversity of Cambridge International Examinations International General Certificate of Secondary Education Biology Paper 1 Multiple Choice October/November 2004 45 Minutesmath magicNo ratings yet
- Algae Anatomy Biochemistry 0Document17 pagesAlgae Anatomy Biochemistry 0Nguyen Hien QuanNo ratings yet
- Nature at Work - The Ongoing Saga of Evolution - 400 PagsDocument400 pagesNature at Work - The Ongoing Saga of Evolution - 400 PagsNoti News WorldNo ratings yet
- Thesis PCRDocument8 pagesThesis PCRgjfs5mtv100% (2)
- 0654 (Biology) ChecklistDocument4 pages0654 (Biology) ChecklistHồ Liên KhảiNo ratings yet
- What Is DNA: FunctionDocument4 pagesWhat Is DNA: FunctionBartoszNo ratings yet
- BSC C501 Biophysics Syllabus - B.SCDocument3 pagesBSC C501 Biophysics Syllabus - B.SC16_dev5038No ratings yet
- Obesity 1. Basic Science of ObesityDocument10 pagesObesity 1. Basic Science of ObesitygraceNo ratings yet
- Photosynthesis AnswersDocument1 pagePhotosynthesis AnswersChristian Hovsep KaradaghlianNo ratings yet
- The Evolution of Bizarre Structures' in Dinosaurs: Biomechanics, Sexual Selection, Social Selection or Species Recognition?Document15 pagesThe Evolution of Bizarre Structures' in Dinosaurs: Biomechanics, Sexual Selection, Social Selection or Species Recognition?Manuel PérezNo ratings yet
- Bla Bla Bla New 2014Document11 pagesBla Bla Bla New 2014Georgiana GeoNo ratings yet
- Cobas Ampliprep Cobas TaqMan HCV Viral LoadDocument84 pagesCobas Ampliprep Cobas TaqMan HCV Viral LoadMolecular_Diagnostics_KKUHNo ratings yet
- Biology Unit 6 Study Guide AnswersDocument3 pagesBiology Unit 6 Study Guide AnswersmisterbrownerNo ratings yet
- Amino Acid StructureDocument26 pagesAmino Acid StructureSaumya SK100% (2)
- CattleDocument66 pagesCattleAlbyziaNo ratings yet
- PCR Troubleshooting-Part 1 "No Bands": Hints, Tips and Trouble Shooting For Molecular Biology TechniciansDocument6 pagesPCR Troubleshooting-Part 1 "No Bands": Hints, Tips and Trouble Shooting For Molecular Biology Technicianssam911101No ratings yet
- Skin Yale University Protein: Where Does Collagen Come From?Document2 pagesSkin Yale University Protein: Where Does Collagen Come From?Ellaine Pearl AlmillaNo ratings yet
- Storing Bacterial Samples For Optimal Viability - Thermo Fisher Scientific - IDDocument3 pagesStoring Bacterial Samples For Optimal Viability - Thermo Fisher Scientific - IDoktaNo ratings yet
- Clinical Genomics Laboratory: Test ResultDocument1 pageClinical Genomics Laboratory: Test ResultPeds Lim PagayatanNo ratings yet
- The Importance of Health Fitness and WellnessDocument3 pagesThe Importance of Health Fitness and Wellnessapi-25954769No ratings yet
- Biodiversity of Flora and FaunaDocument13 pagesBiodiversity of Flora and FaunaLasmaenita SiahaanNo ratings yet
- General Biology 1 Summative Assessment Week 1-4Document2 pagesGeneral Biology 1 Summative Assessment Week 1-4Cherrina Aguila100% (1)
- (José Das Neves, Bruno Sarmento (Eds.) ) Mucosal DDocument603 pages(José Das Neves, Bruno Sarmento (Eds.) ) Mucosal Dfaysal100% (1)
- Pneumacult™-Ali: For Bronchial Epithelial CellsDocument2 pagesPneumacult™-Ali: For Bronchial Epithelial CellsDunkMeNo ratings yet
- XXXDocument134 pagesXXXrhlzuraNo ratings yet
- Company. PP 135-152.: ReferencesDocument10 pagesCompany. PP 135-152.: ReferencesGlascow universityNo ratings yet