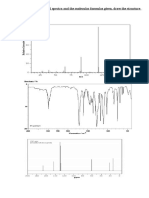
You might also like
- MRI Study GuideDocument66 pagesMRI Study GuideJuan Doval100% (7)
- Infrared SpectrosDocument59 pagesInfrared SpectrosMohammed Muzaffar MoinuddinNo ratings yet
- PMR Spectroscopy: Solved Problems Volume : IIFrom EverandPMR Spectroscopy: Solved Problems Volume : IIRating: 5 out of 5 stars5/5 (3)
- Atkins, Solution, 7th EdDocument21 pagesAtkins, Solution, 7th Edapi-3723327100% (3)
- Basics of MRI PDFDocument44 pagesBasics of MRI PDFBMT50% (2)
- NMR SpectrosDocument66 pagesNMR SpectrosDushyant Patel100% (1)
- Resonance Raman SpectrosDocument14 pagesResonance Raman SpectrosSarthak Mandal0% (1)
- Spin-Spin Coupling in NMRDocument17 pagesSpin-Spin Coupling in NMRBenjamín Marc Ridgway de SassouNo ratings yet
- Mass SpectrosDocument43 pagesMass Spectrosapi-3723327100% (4)
- NMR Workshop ReportDocument18 pagesNMR Workshop ReportmrtharamNo ratings yet
- Molecular Spectroscopy: BackgroundDocument45 pagesMolecular Spectroscopy: Backgroundsavvy_as_98100% (1)
- Basics in NMRDocument75 pagesBasics in NMRPrashant PandeyNo ratings yet
- Reaction Mechanisms GOC BookDocument84 pagesReaction Mechanisms GOC BookAyushNo ratings yet
- CHEM 430 NMR Spectroscopy Chapter 3Document97 pagesCHEM 430 NMR Spectroscopy Chapter 3Praveen kumar100% (1)
- Atomic Emission SpectrosDocument5 pagesAtomic Emission SpectrosBea Uy0% (1)
- (UV Vis) SpectrosDocument4 pages(UV Vis) SpectrosGarion Charles0% (1)
- Mass Spectrometry 1Document33 pagesMass Spectrometry 1PhiPhiNo ratings yet
- IR SpectrosDocument41 pagesIR SpectrosKD LoteyNo ratings yet
- HPLC Mpat Sem1Document28 pagesHPLC Mpat Sem1RAKSHA ARUNNo ratings yet
- Lecture 5 Molecular SpectrosDocument97 pagesLecture 5 Molecular Spectroshoboslayer97No ratings yet
- Isolation Purification and Identification of CurcuminoidsDocument5 pagesIsolation Purification and Identification of CurcuminoidsNguyenVan HanNo ratings yet
- NMR Lecture FinalDocument98 pagesNMR Lecture FinalXarOonNo ratings yet
- Chapter 5 Principle and Application of UV and Visible SpectrosDocument24 pagesChapter 5 Principle and Application of UV and Visible SpectrosEsuendalew DebebeNo ratings yet
- Submitted By: Ms - Bushra Qamar Ms63-10-815Document46 pagesSubmitted By: Ms - Bushra Qamar Ms63-10-815Faazi'z Pari100% (1)
- Introduction To Infrared Spectroscopy: D. Jim Livingston Faculty of Chemistry St. John'S CollegeDocument22 pagesIntroduction To Infrared Spectroscopy: D. Jim Livingston Faculty of Chemistry St. John'S CollegeJim LivingstonNo ratings yet
- Presentation Uv SpectrophotometerDocument14 pagesPresentation Uv SpectrophotometerKrishna DabgarNo ratings yet
- NMR Solving StrategyDocument2 pagesNMR Solving Strategysorrow Lemon100% (1)
- 13C NMR SpectrosDocument16 pages13C NMR Spectrosapi-3723327100% (4)
- Chapter 19 NMRDocument126 pagesChapter 19 NMRMuchammad RofiiNo ratings yet
- Spectroscopic Solutions of StructureDocument21 pagesSpectroscopic Solutions of StructureKassimNo ratings yet
- Chapter 2 Infrared Spectroscopy IRDocument28 pagesChapter 2 Infrared Spectroscopy IRAmeerRashidNo ratings yet
- Guide To Solving Spectroscopy ProblemsDocument4 pagesGuide To Solving Spectroscopy ProblemsJen100% (1)
- Various Tools and Techniques Used For Structural ElucidationDocument28 pagesVarious Tools and Techniques Used For Structural ElucidationAvinashNo ratings yet
- Uv Visible Spectroscopy: by Nandesh V. PingaleDocument38 pagesUv Visible Spectroscopy: by Nandesh V. PingaleMohammed Adil ShareefNo ratings yet
- Iub Pha404 Autumn 2022 Ms BasicDocument52 pagesIub Pha404 Autumn 2022 Ms BasicTanvir FahimNo ratings yet
- Mass Spectrometry InstrumentationDocument3 pagesMass Spectrometry InstrumentationSangeetha priya SNo ratings yet
- AromaticityDocument12 pagesAromaticityV G Viju KumarNo ratings yet
- Electronic SpectrosDocument82 pagesElectronic SpectrosEub EuNo ratings yet
- Report NMRDocument13 pagesReport NMRsarahNo ratings yet
- Raman SpectrosDocument15 pagesRaman Spectroscarlosev11No ratings yet
- Sn2 and Sn1 Reactions: Sethuranga Skanthan.P.TDocument12 pagesSn2 and Sn1 Reactions: Sethuranga Skanthan.P.TSethuNo ratings yet
- Infrared SpectrosDocument4 pagesInfrared Spectrosjellybean07100% (1)
- Chapter 7Document101 pagesChapter 7Asinake AlemayehuNo ratings yet
- Proton Nuclear Magnetic Resonance Spectroscopy H - NMRDocument61 pagesProton Nuclear Magnetic Resonance Spectroscopy H - NMRchemist100% (2)
- UV Spectroscopy 2016Document87 pagesUV Spectroscopy 2016M Mudassar AslamNo ratings yet
- Mass SpectrometryDocument52 pagesMass Spectrometrybbhavya50% (2)
- Analytical ChemistryDocument14 pagesAnalytical ChemistryDrMd Idris100% (2)
- Decoupling: Homonuclear and HeteronuclearDocument7 pagesDecoupling: Homonuclear and HeteronuclearDaniela M. ZanataNo ratings yet
- IR SpectrosDocument27 pagesIR SpectrosAlyssa NesianandaNo ratings yet
- FMO LectureDocument14 pagesFMO Lecturebooks4free23No ratings yet
- Raman Spectroscopy: Dr. Majid Muneer Assistant Professor Department of Chemistry, GCU FaisalabadDocument47 pagesRaman Spectroscopy: Dr. Majid Muneer Assistant Professor Department of Chemistry, GCU FaisalabadMartyr LeoNo ratings yet
- 2D NMRlatestDocument34 pages2D NMRlatestNandan ShindeNo ratings yet
- ChromatographyDocument19 pagesChromatographyM.PRASAD NAIDU0% (1)
- The Raman EffectDocument12 pagesThe Raman Effectmkbh10No ratings yet
- Mass Spectrometry: Ev MVDocument12 pagesMass Spectrometry: Ev MVMuhammad Tariq RazaNo ratings yet
- NMR Spectroscopy-1Document63 pagesNMR Spectroscopy-1Abhishek Kumar SinghNo ratings yet
- IR SpectrosDocument119 pagesIR SpectrosRojan PradhanNo ratings yet
- Mass Spectroscopy: - : Presented By:-Amruta S. Sambarekar 1 Year M.Pharm Dept. of Pharmaceutics M M C P, BelgaumDocument56 pagesMass Spectroscopy: - : Presented By:-Amruta S. Sambarekar 1 Year M.Pharm Dept. of Pharmaceutics M M C P, BelgaumSudeep_Kolhar_5702No ratings yet
- Infrared Spectros PDFDocument33 pagesInfrared Spectros PDFMuhammad BilalNo ratings yet
- RamanDocument29 pagesRamanapi-3723327100% (7)
- Optical Instrum 1Document11 pagesOptical Instrum 1api-3723327No ratings yet
- Optical Instrum (Net)Document23 pagesOptical Instrum (Net)api-3723327100% (2)
- IRDocument27 pagesIRapi-3723327No ratings yet
- Mass SpectDocument28 pagesMass Spectapi-3723327100% (1)
- Concpts Txcity FiguresDocument21 pagesConcpts Txcity Figuresapi-3723327No ratings yet
- Bio Degradation of PollutantsDocument40 pagesBio Degradation of Pollutantsapi-3723327No ratings yet
- 13C NMR SpectrosDocument16 pages13C NMR Spectrosapi-3723327100% (4)
- Chap TiDocument76 pagesChap Tiapi-3723327No ratings yet
- Oxidative StressDocument53 pagesOxidative Stressapi-3723327100% (2)
- Atkins, Solution, 7th EdDocument22 pagesAtkins, Solution, 7th Edapi-3723327No ratings yet
- Atkins, Solution, 7th EdDocument15 pagesAtkins, Solution, 7th Edapi-3723327No ratings yet
- Atkins, Solution, 7th EdDocument10 pagesAtkins, Solution, 7th Edapi-3723327No ratings yet
- Atkins, Solution, 7th EdDocument12 pagesAtkins, Solution, 7th Edapi-3723327No ratings yet
- Atkins, Solution, 7th EdDocument14 pagesAtkins, Solution, 7th Edapi-3723327No ratings yet
- Atkins, Solution, 7th EdDocument11 pagesAtkins, Solution, 7th Edapi-3723327No ratings yet
- Atkins, Solution, 7th EdDocument19 pagesAtkins, Solution, 7th Edapi-3723327No ratings yet
- Magnets, Spins, and Resonances - An Introduction To The Basics of Magnetic Resonance SAPEDM Magnete Spins Resonanzen MR-07000G.643.02.02.02Document244 pagesMagnets, Spins, and Resonances - An Introduction To The Basics of Magnetic Resonance SAPEDM Magnete Spins Resonanzen MR-07000G.643.02.02.02Thamiris RosaNo ratings yet
- Quantitative 1H NMR SpectrosDocument22 pagesQuantitative 1H NMR Spectrossantosh0912830% (1)
- Akitt, J. W. - Mann, Brian E - NMR and Chemistry - An Introduction To Modern NMR Spectroscopy, Fourth Edition-CRC Press (2017)Document417 pagesAkitt, J. W. - Mann, Brian E - NMR and Chemistry - An Introduction To Modern NMR Spectroscopy, Fourth Edition-CRC Press (2017)joaoDjmsNo ratings yet
- Magnetic Resonance of Semiconductors and Their Nanostructures Basic and Advanced ApplicationsDocument535 pagesMagnetic Resonance of Semiconductors and Their Nanostructures Basic and Advanced ApplicationsAlbertoPitaNo ratings yet
- The Potential of Nuclear Magnetic Resonance (NMR) To Non-Destructively Characterize Early-Age Concrete by An One-Sided Access (OSA) TechniqueDocument10 pagesThe Potential of Nuclear Magnetic Resonance (NMR) To Non-Destructively Characterize Early-Age Concrete by An One-Sided Access (OSA) TechniqueBabak SalimifardNo ratings yet
- Cyclen Based GD ComplexesDocument22 pagesCyclen Based GD ComplexesHaroon Ur RashidNo ratings yet
- ACERT Workshop - Overview of EPR Methods To Determine Interspin DistancesDocument47 pagesACERT Workshop - Overview of EPR Methods To Determine Interspin DistancesChung-Ta HanNo ratings yet
- Basics of Measuring The Dielectric Properties of MaterialsDocument16 pagesBasics of Measuring The Dielectric Properties of MaterialsYouli TianNo ratings yet
- Spwla 2015 Formation Logging PDFDocument11 pagesSpwla 2015 Formation Logging PDFDaniel nasasiraNo ratings yet
- ENCHighlightsDocument60 pagesENCHighlightsUtpal SharmaNo ratings yet
- Course No. Course Name Credits: Medicinal ChemistryDocument10 pagesCourse No. Course Name Credits: Medicinal ChemistryHeena BhojwaniNo ratings yet
- Chapter 01 Introduction Qubit DecoherenceDocument49 pagesChapter 01 Introduction Qubit DecoherenceValentinaNo ratings yet
- Schlumberger Log Interpretation ChartsDocument287 pagesSchlumberger Log Interpretation Chartsice_PL100% (10)
- CBM355 Medical Imaging Systems 2-MarksDocument23 pagesCBM355 Medical Imaging Systems 2-Markssanthosh sekar100% (2)
- Presymposium Workshop Part2 May28Document71 pagesPresymposium Workshop Part2 May28Smruti Snigdha MishraNo ratings yet
- Principles of NMR SpectrosDocument23 pagesPrinciples of NMR SpectrosSudha AtmakuriNo ratings yet
- Halse Centric Scan SPRITE MRIDocument11 pagesHalse Centric Scan SPRITE MRIAlfonso LemaNo ratings yet
- The Preparation of Magnetic Nanoparticles For Applications in BiomedicineDocument17 pagesThe Preparation of Magnetic Nanoparticles For Applications in BiomedicineMadavat Prem Pai LópezNo ratings yet
- Polymer SpectrosDocument408 pagesPolymer SpectrosIkbal Fikri100% (3)
- Qcadesigner SimuDocument20 pagesQcadesigner SimuSupravatAppaNo ratings yet
- Basic Physical Principles: of MRIDocument35 pagesBasic Physical Principles: of MRInaveeninsaneNo ratings yet
- My Summary in Radiology - A M Abodahab MDDocument387 pagesMy Summary in Radiology - A M Abodahab MDtran quang vinh100% (1)
- Ab Initio Hydrogen-Vacancy FormaionDocument10 pagesAb Initio Hydrogen-Vacancy FormaionWang ShuaiNo ratings yet
- Biomedical Magnetic Resonance:: Proceedings of The International WorkshopDocument475 pagesBiomedical Magnetic Resonance:: Proceedings of The International Workshopsalah subbah0% (1)
- MRI MohemDocument7 pagesMRI Mohema.mahdieh90No ratings yet
- NMR Lecture SOSDocument43 pagesNMR Lecture SOSpoornanandhanNo ratings yet
- NMR Logging Activation Sets Selection and Fluid Relaxation CH - 2018 - Natural GDocument7 pagesNMR Logging Activation Sets Selection and Fluid Relaxation CH - 2018 - Natural GMinh Hong PhamNo ratings yet
- Bose Einstein Liquid Helium PDFDocument9 pagesBose Einstein Liquid Helium PDFKevin Omar Alvarado VegaNo ratings yet