You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Exam Primer McqsDocument25 pagesExam Primer Mcqsjohn eric100% (1)
- Susskind Quantum Mechanics NotesDocument27 pagesSusskind Quantum Mechanics NotesGary SheaNo ratings yet
- Christopher D. Paulse and Jonathan Tennyson - An Empirical Potential Energy Surface For Water Accounting For States With High Angular MomentumDocument10 pagesChristopher D. Paulse and Jonathan Tennyson - An Empirical Potential Energy Surface For Water Accounting For States With High Angular MomentumPassammNo ratings yet
- Jonathan Tennyson and Steven Miller - H3 +: From First Principles To JupiterDocument12 pagesJonathan Tennyson and Steven Miller - H3 +: From First Principles To JupiterPassammNo ratings yet
- C. Ruth Le. Sueur, James R. Henderson and Jonathan Tennyson - Gateway States and Bath States in The Vibrational Spectrum of H3 +Document8 pagesC. Ruth Le. Sueur, James R. Henderson and Jonathan Tennyson - Gateway States and Bath States in The Vibrational Spectrum of H3 +PassammNo ratings yet
- Jonathan Tennyson, Steven Miller and C. Ruth Le Sueur - TRIATOM: Programs For The Calculation of Ro-Vibrational Spectra of Triatomic MoleculesDocument26 pagesJonathan Tennyson, Steven Miller and C. Ruth Le Sueur - TRIATOM: Programs For The Calculation of Ro-Vibrational Spectra of Triatomic MoleculesPassammNo ratings yet
- M. E. Bannister Et Al - Absolute Cross Sections For Near-Threshold Electron-Impact Excitation of The 2s 2S - 2p 2 P Transition in C 3+Document13 pagesM. E. Bannister Et Al - Absolute Cross Sections For Near-Threshold Electron-Impact Excitation of The 2s 2S - 2p 2 P Transition in C 3+PassammNo ratings yet
- James R. Henderson Et Al - Coordinate Ordering in The Discrete Variable RepresentationDocument6 pagesJames R. Henderson Et Al - Coordinate Ordering in The Discrete Variable RepresentationPassammNo ratings yet
- Baljit K. Sarpal and Jonathan Tennyson - Calculated Vibrational Excitation Rates For Electron - H2 + CollisionsDocument4 pagesBaljit K. Sarpal and Jonathan Tennyson - Calculated Vibrational Excitation Rates For Electron - H2 + CollisionsPassammNo ratings yet
- Gilda E. Ballester Et Al - Latitudinal Temperature Variations of Jovian H3 +Document6 pagesGilda E. Ballester Et Al - Latitudinal Temperature Variations of Jovian H3 +PassammNo ratings yet
- Bianca M. Dinelli Et Al - A Spectroscopically Determined Potential Energy Surface For H3 +Document9 pagesBianca M. Dinelli Et Al - A Spectroscopically Determined Potential Energy Surface For H3 +PassammNo ratings yet
- Jonathan Tennyson, Steven Miller and Hans Schild - First-Principles Calculations On The Astrochemistry and Spectroscopy of H3 +Document5 pagesJonathan Tennyson, Steven Miller and Hans Schild - First-Principles Calculations On The Astrochemistry and Spectroscopy of H3 +PassammNo ratings yet
- James R. Henderson and Jonathan Tennyson - DVR1D: Programs For Mixed Pointwise/ Basis Set Calculation of Ro-Vibrational SpectraDocument14 pagesJames R. Henderson and Jonathan Tennyson - DVR1D: Programs For Mixed Pointwise/ Basis Set Calculation of Ro-Vibrational SpectraPassammNo ratings yet
- James R. Henderson, C. Ruth Le Sueur and Jonathan Tennyson - DVR3D: Programs For Fully Pointwise Calculation of Vibrational SpectraDocument17 pagesJames R. Henderson, C. Ruth Le Sueur and Jonathan Tennyson - DVR3D: Programs For Fully Pointwise Calculation of Vibrational SpectraPassammNo ratings yet
- Oleg L. Polyanksy Et Al - Rotational Levels of H2D +: Variational Calculations and AssignmentsDocument11 pagesOleg L. Polyanksy Et Al - Rotational Levels of H2D +: Variational Calculations and AssignmentsPassammNo ratings yet
- L.M. Trafton Et Al - Detection of H3 + From UranusDocument6 pagesL.M. Trafton Et Al - Detection of H3 + From UranusPassammNo ratings yet
- Oleg L. Polyansky and Jonathan Tennyson - On The Convergence of Effective Hamiltonian ExpansionsDocument6 pagesOleg L. Polyansky and Jonathan Tennyson - On The Convergence of Effective Hamiltonian ExpansionsPassammNo ratings yet
- C. Ruth Le Sueuer Et Al - On The Use of Variational Wavefunctions in Calculating Vibrational Band IntensitiesDocument10 pagesC. Ruth Le Sueuer Et Al - On The Use of Variational Wavefunctions in Calculating Vibrational Band IntensitiesPassammNo ratings yet
- Bianca M. Dinelli Et Al - Bands of H3 + Up To 4-Nu-2 Rovibrational Transitions From First Principles CalculationsDocument8 pagesBianca M. Dinelli Et Al - Bands of H3 + Up To 4-Nu-2 Rovibrational Transitions From First Principles CalculationsPassammNo ratings yet
- Jonathan Tennyson - Why Calculate The Spectra of Small Molecules?Document9 pagesJonathan Tennyson - Why Calculate The Spectra of Small Molecules?PassammNo ratings yet
- James R. Henderson, Hoanh A. Lam and Jonathan Tennyson - Highly Excited Vibrational States of The KCN MoleculeDocument7 pagesJames R. Henderson, Hoanh A. Lam and Jonathan Tennyson - Highly Excited Vibrational States of The KCN MoleculePassammNo ratings yet
- Steven Miller and Jonathan Tennyson - H3 + in SpaceDocument8 pagesSteven Miller and Jonathan Tennyson - H3 + in SpacePassammNo ratings yet
- Discretization To Avoid Singularities in Vibration-Rotation Hamiltonians: Bisector Embedding For AB2 TriatomicsDocument12 pagesDiscretization To Avoid Singularities in Vibration-Rotation Hamiltonians: Bisector Embedding For AB2 TriatomicsPassammNo ratings yet
- Michael Berblinger and Christoph Schlier - Accurate Specific Molecular State Densities by Phase Space Integration. I. Computational MethodDocument9 pagesMichael Berblinger and Christoph Schlier - Accurate Specific Molecular State Densities by Phase Space Integration. I. Computational MethodPassammNo ratings yet
- John A. Fernley Et Al - Band Origins For Water Up To 22 000 CM - 1: A Comparison of Spectroscopically Determined Potential Energy SurfacesDocument13 pagesJohn A. Fernley Et Al - Band Origins For Water Up To 22 000 CM - 1: A Comparison of Spectroscopically Determined Potential Energy SurfacesPassammNo ratings yet
- Steven Miller, Jonathan Tennyson and John Fernley - Calculation of Transition Frequencies and Line Strengths of Water For Cool Star OpacitiesDocument8 pagesSteven Miller, Jonathan Tennyson and John Fernley - Calculation of Transition Frequencies and Line Strengths of Water For Cool Star OpacitiesPassammNo ratings yet
- K.S. Sidhu Et Al - Partition Functions and Equilibrium Constants For H3 + and H2D +Document4 pagesK.S. Sidhu Et Al - Partition Functions and Equilibrium Constants For H3 + and H2D +PassammNo ratings yet
- Shih-I Chu - Rotational Excitation of Symmetric-Top Molecular Ions by Electron ImpactDocument10 pagesShih-I Chu - Rotational Excitation of Symmetric-Top Molecular Ions by Electron ImpactPassammNo ratings yet
- Jonathan Tennyson and Steven Miller - Calculating Molecular SpectraDocument9 pagesJonathan Tennyson and Steven Miller - Calculating Molecular SpectraPassammNo ratings yet
- Steven Miller Et Al - Identification of Features Due To H3 + in The Infrared Spectrum of Supernova 1987ADocument3 pagesSteven Miller Et Al - Identification of Features Due To H3 + in The Infrared Spectrum of Supernova 1987APassammNo ratings yet
- Lancelot Kao Et Al - A Table of Astronomically Important Ro-Vibrational Transitions For The H3 + Molecular IonDocument13 pagesLancelot Kao Et Al - A Table of Astronomically Important Ro-Vibrational Transitions For The H3 + Molecular IonPassammNo ratings yet
- Viatcheslav Kokoouline Et Al - Calculation of Rate Constants For Vibrational and Rotational Excitation of The H3 + Ion by Electron ImpactDocument8 pagesViatcheslav Kokoouline Et Al - Calculation of Rate Constants For Vibrational and Rotational Excitation of The H3 + Ion by Electron ImpactPassammNo ratings yet
- EPFL Lectures On Conformal Field Theory in D 3 DimensionsDocument68 pagesEPFL Lectures On Conformal Field Theory in D 3 DimensionsMojeime Igor NowakNo ratings yet
- CFD Abl2Document38 pagesCFD Abl2Rahul ShuklaNo ratings yet
- Physics Lecture Notes Elasticity Stress Strain ModuliDocument18 pagesPhysics Lecture Notes Elasticity Stress Strain ModuliMarian Galvez-LuisNo ratings yet
- FIITJEE ALL INDIA TEST SERIES PART TEST – I PHYSICS ANSWERS, HINTS & SOLUTIONSDocument18 pagesFIITJEE ALL INDIA TEST SERIES PART TEST – I PHYSICS ANSWERS, HINTS & SOLUTIONSShiromani BujdilNo ratings yet
- Exercise 7: Tying A Well To A Seismic Line: ObjectiveDocument5 pagesExercise 7: Tying A Well To A Seismic Line: ObjectiveSadiq JalalNo ratings yet
- Tutorial on Gas, Liquid and Solid DielectricsDocument5 pagesTutorial on Gas, Liquid and Solid DielectricsWeiJin ChaiNo ratings yet
- 651 PDFDocument12 pages651 PDFjitendraNo ratings yet
- Learning What Makes Things MoveDocument7 pagesLearning What Makes Things MoveAnubhuti GhaiNo ratings yet
- Process Dynamics and Control: By: Gemechu Bushu (AAU)Document32 pagesProcess Dynamics and Control: By: Gemechu Bushu (AAU)gebreslassie gereziherNo ratings yet
- NET December 2016 Physics Exam QuestionsDocument21 pagesNET December 2016 Physics Exam QuestionsRamesh IswaraNo ratings yet
- CCHE4271: Preliminary Examination in ChemistryDocument8 pagesCCHE4271: Preliminary Examination in ChemistryEkanisaKurniawatiNo ratings yet
- TikZ-Feynman: Draw Feynman Diagrams with TikZDocument33 pagesTikZ-Feynman: Draw Feynman Diagrams with TikZwintersoulsNo ratings yet
- Acceptance Angle and Numerical ApertureDocument2 pagesAcceptance Angle and Numerical ApertureAbhishek BhowmikNo ratings yet
- Mechanical Design of Overhead LinesDocument9 pagesMechanical Design of Overhead Linesmuhammad_sarwar_27No ratings yet
- 6 Some Important Features of Stone Column TreatmentDocument7 pages6 Some Important Features of Stone Column TreatmentBilal Ahmed BarbhuiyaNo ratings yet
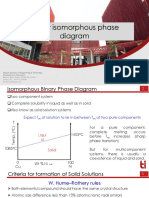
- Binary Phase Diagram Key DetailsDocument13 pagesBinary Phase Diagram Key DetailsRohanNo ratings yet
- RotameterDocument8 pagesRotameterBabylyn AustriaNo ratings yet
- Applied Thermal Engineering: SciencedirectDocument15 pagesApplied Thermal Engineering: SciencedirectSadegh AhmadiNo ratings yet
- Wood 1902Document9 pagesWood 1902Lucho GonzalezNo ratings yet
- Paribhashik ShabdawaliDocument5 pagesParibhashik Shabdawalisgangwar2005sg100% (1)
- Classxiiassignment201920Document84 pagesClassxiiassignment201920Dhaval KumarNo ratings yet
- Doppler Effect Sound Frequency ChangesDocument10 pagesDoppler Effect Sound Frequency ChangesfizznuarNo ratings yet
- MicromagnetsDocument5 pagesMicromagnetsTa BarNo ratings yet
- Chapter-4 - Moving Charges and MagnetismDocument8 pagesChapter-4 - Moving Charges and MagnetismAdarsh SilkotiNo ratings yet
- 7 - GravitationDocument5 pages7 - GravitationAvik DasNo ratings yet
- Significant Figures, Scientific Notation and Metric PrefixesDocument3 pagesSignificant Figures, Scientific Notation and Metric PrefixesmphoNo ratings yet
- Xvii Paper 44Document17 pagesXvii Paper 44dhavaleshNo ratings yet
- BE 8256 - Basic Mechanical Engineering / U – II / Compiled by R.Arul KamalakumarDocument24 pagesBE 8256 - Basic Mechanical Engineering / U – II / Compiled by R.Arul KamalakumararulrakkNo ratings yet