You might also like
- PMR Spectroscopy: Solved Problems Volume : IIFrom EverandPMR Spectroscopy: Solved Problems Volume : IIRating: 5 out of 5 stars5/5 (3)
- Absorption Spectra and Chemical Bonding in ComplexesFrom EverandAbsorption Spectra and Chemical Bonding in ComplexesRating: 2.5 out of 5 stars2.5/5 (2)
- Spectroscopic Solutions of StructureDocument21 pagesSpectroscopic Solutions of StructureKassimNo ratings yet
- Guide To Solving Spectroscopy ProblemsDocument4 pagesGuide To Solving Spectroscopy ProblemsJen100% (1)
- Experiment 2. Separation of Compounds by Paper ChromatographyDocument11 pagesExperiment 2. Separation of Compounds by Paper ChromatographybidinNo ratings yet
- NMR Solving StrategyDocument2 pagesNMR Solving Strategysorrow Lemon100% (1)
- AC 101 Unit 1 Titrimetric AnalysisDocument90 pagesAC 101 Unit 1 Titrimetric AnalysisRishabh Kumar Singh100% (1)
- Aromaticity CompleteDocument104 pagesAromaticity Completewahidalwahdi100% (1)
- 1 IR NMR Practice ProblemsetDocument12 pages1 IR NMR Practice ProblemsetJustin BuiNo ratings yet
- Molecular RearrangementsDocument9 pagesMolecular RearrangementsDhanaswamy Ilangeswaran67% (3)
- Module8 PDFDocument40 pagesModule8 PDFFaizan AhmadNo ratings yet
- Kuliah MG 9 Racemix Mixture and ResolutionDocument184 pagesKuliah MG 9 Racemix Mixture and ResolutionBowo Aank ApriantoNo ratings yet
- Silverstein Chapter 1 Mass SpectrometryDocument71 pagesSilverstein Chapter 1 Mass SpectrometryNikita GroverNo ratings yet
- AromaticityDocument12 pagesAromaticityV G Viju KumarNo ratings yet
- Molecular Spectroscopy: BackgroundDocument45 pagesMolecular Spectroscopy: Backgroundsavvy_as_98100% (1)
- NMR SpectrosDocument185 pagesNMR SpectrosBathir JafarNo ratings yet
- (UV Vis) SpectrosDocument4 pages(UV Vis) SpectrosGarion Charles0% (1)
- Huckel Theory For Conjugated Systems: CH 105: Organic ChemistryDocument72 pagesHuckel Theory For Conjugated Systems: CH 105: Organic ChemistryRaunaq Bhirangi100% (1)
- Retrosynthetic Analysis PDFDocument6 pagesRetrosynthetic Analysis PDFNoleNo ratings yet
- Clusters and Catenation in P-Block: Allotropes of CarbonDocument15 pagesClusters and Catenation in P-Block: Allotropes of Carbonrajender kumarNo ratings yet
- Lecture Notes 2 Nano MaterialsDocument21 pagesLecture Notes 2 Nano MaterialsHuzaifa ShabbirNo ratings yet
- Mass Spectra Worksheet 1Document5 pagesMass Spectra Worksheet 1scribdfreepdfNo ratings yet
- Elimination ReactionsDocument7 pagesElimination ReactionsIrfan IslamyNo ratings yet
- Practice Problems For Physical Chemistry 2Document1 pagePractice Problems For Physical Chemistry 2Fatima CellonaNo ratings yet
- Aromaticity NotesDocument10 pagesAromaticity NotesVirendra Singh Rajput100% (1)
- Spec Prob Set 315 CurrentDocument20 pagesSpec Prob Set 315 CurrentUmang Agarwal57% (7)
- 13C NMR SpectrosDocument16 pages13C NMR Spectrosapi-3723327100% (4)
- Metal Complexes or Coordination Compounds: Kfecn 4K Fe CNDocument90 pagesMetal Complexes or Coordination Compounds: Kfecn 4K Fe CNPavan Boro100% (1)
- The Transition Metals, The Lanthanides and The AntinidesDocument21 pagesThe Transition Metals, The Lanthanides and The AntinidesApril CruzNo ratings yet
- Chap21 Skoog PotentiometryDocument55 pagesChap21 Skoog PotentiometryMarielle Perejon100% (1)
- Organometallic Chemistry: CH 431 MFT CH 13Document37 pagesOrganometallic Chemistry: CH 431 MFT CH 13Vaittianathan MahavapillaiNo ratings yet
- MOT UploadDocument36 pagesMOT UploadSarthak Singh100% (1)
- 01 GC TheoryDocument71 pages01 GC Theory03ASEPJAELANINo ratings yet
- Report NMRDocument13 pagesReport NMRsarahNo ratings yet
- FMO LectureDocument14 pagesFMO Lecturebooks4free23No ratings yet
- Questions On StereochemistryDocument2 pagesQuestions On StereochemistryShilajit BaruaNo ratings yet
- UV Spectroscopy 2016Document87 pagesUV Spectroscopy 2016M Mudassar AslamNo ratings yet
- Organometal Chem PDFDocument55 pagesOrganometal Chem PDFSushmita Dey100% (1)
- IR - HNMR ProblemsDocument33 pagesIR - HNMR Problemsbsakaly112100% (1)
- BW Mass Spectrometry - ZeeshanDocument59 pagesBW Mass Spectrometry - ZeeshanAdnan RoonjhaNo ratings yet
- NMR Practice ProblemsDocument9 pagesNMR Practice ProblemsVivek AgrahariNo ratings yet
- SET-NET Pericyclic ReactionsDocument61 pagesSET-NET Pericyclic ReactionsBapu ThoratNo ratings yet
- Organic Structure Elucidation WorkbookDocument498 pagesOrganic Structure Elucidation WorkbookKajan Muneeswaran75% (4)
- Lucas Test PDFDocument3 pagesLucas Test PDFciciNo ratings yet
- NMR SpectrosDocument29 pagesNMR Spectroshareesh13h100% (1)
- CH 431 Lab ManualFullDocument28 pagesCH 431 Lab ManualFullHân BảoNo ratings yet
- Lab #1: Absorption Spectra of Conjugated Dyes: E E E EDocument5 pagesLab #1: Absorption Spectra of Conjugated Dyes: E E E EIreneVeladoNo ratings yet
- Isolobal AnalogyDocument4 pagesIsolobal Analogyindu priyaNo ratings yet
- Journal of Electroanalytical Chemistry 609 (2007) 17-26Document10 pagesJournal of Electroanalytical Chemistry 609 (2007) 17-26Alex B-RomeroNo ratings yet
- Gravimetric Analysis and Precipitation - TitrationsDocument34 pagesGravimetric Analysis and Precipitation - TitrationsElvinNo ratings yet
- Electrochemistry Exercise SolutionDocument22 pagesElectrochemistry Exercise SolutionGOURISH AGRAWALNo ratings yet
- NMR HandoutDocument23 pagesNMR HandoutVirendra Singh RajputNo ratings yet
- 3820 Lecture Chapter - 3 - Part1 - 2004 PDFDocument15 pages3820 Lecture Chapter - 3 - Part1 - 2004 PDFPhuongUblMyNo ratings yet
- CAIE Chemistry A-Level: 24: ElectrochemistryDocument8 pagesCAIE Chemistry A-Level: 24: ElectrochemistryahumanbeinginearthNo ratings yet
- Isolation Purification and Identification of CurcuminoidsDocument5 pagesIsolation Purification and Identification of CurcuminoidsNguyenVan HanNo ratings yet
- NMR 1Document49 pagesNMR 1Jyoti ChaturvediNo ratings yet
- Aldehydes and KetonesDocument5 pagesAldehydes and KetonesBaji Babu BejjankiNo ratings yet
- Chemistry II (Organic) Heteroaromatic Chemistry Lectures 2 & 3Document25 pagesChemistry II (Organic) Heteroaromatic Chemistry Lectures 2 & 3Subhabrata MabhaiNo ratings yet
- GRE Sub 化学题 (太傻整理)Document30 pagesGRE Sub 化学题 (太傻整理)Alisa100% (1)
- Experiment 1 Solubility of Organic CompoundsDocument3 pagesExperiment 1 Solubility of Organic CompoundsIshaa IluminNo ratings yet
- Organic Compounds: Are They Useful?: Science 9 Week 4 WorksheetsDocument4 pagesOrganic Compounds: Are They Useful?: Science 9 Week 4 WorksheetsGEROME REY LINONo ratings yet
- PYQ of Organic Nomenclature NEET 2022Document25 pagesPYQ of Organic Nomenclature NEET 2022Saloni tyagi100% (2)
- 26 Halogen Derivatives Formula Sheets Getmarks AppDocument10 pages26 Halogen Derivatives Formula Sheets Getmarks AppsinghrishxbhNo ratings yet
- 11 Alcohols Phenols and EthersDocument6 pages11 Alcohols Phenols and EthersVansh VaibhavNo ratings yet
- Heterocycles Essentials1-2009Document2 pagesHeterocycles Essentials1-2009Aravindan NatarajanNo ratings yet
- ReasoningDocument4 pagesReasoningAayush MishraNo ratings yet
- Organic Chemistry - Chapter 19 - NitrilesDocument5 pagesOrganic Chemistry - Chapter 19 - NitrilesSairille ManejaNo ratings yet
- ORGANIC Chemistry: 1. S.No. Compound Aromatic Anti-Aromatic Non-AromaticDocument6 pagesORGANIC Chemistry: 1. S.No. Compound Aromatic Anti-Aromatic Non-AromaticTarun SoniNo ratings yet
- Alcohols & Ethers Exercise - IDocument5 pagesAlcohols & Ethers Exercise - IVasudev ArchakNo ratings yet
- Recognizing Endo and Exo - Master Organic ChemistryDocument9 pagesRecognizing Endo and Exo - Master Organic ChemistryashishNo ratings yet
- Hydrocarbons QuestionsDocument5 pagesHydrocarbons QuestionsBhakti Nath MishraNo ratings yet
- Alkynes 1Document27 pagesAlkynes 1Irfan GumelarNo ratings yet
- Al KanesDocument35 pagesAl Kanessimonatics08No ratings yet
- Chemistry Xii NAME: - : Alcohol, Phenol & EtherDocument1 pageChemistry Xii NAME: - : Alcohol, Phenol & EtherSahir Hemnani100% (1)
- Chapter 24 - Organic ChemistryDocument13 pagesChapter 24 - Organic Chemistrymaniz442No ratings yet
- Organic Chemistry: Dr. Omar Mohammed YahyaDocument15 pagesOrganic Chemistry: Dr. Omar Mohammed YahyaCover SongsNo ratings yet
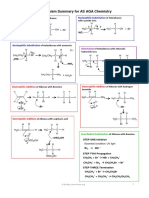
- Mechanism Summary For AS AQA Chemistry: HO: NCDocument4 pagesMechanism Summary For AS AQA Chemistry: HO: NCjohn mNo ratings yet
- T12 Introduction To Organic Chemistry 27-34Document8 pagesT12 Introduction To Organic Chemistry 27-34饶宝珍No ratings yet
- Lecture 2 - S1 - 96 PDFDocument15 pagesLecture 2 - S1 - 96 PDFAnonymous dSQiRGNo ratings yet
- Transition Metal CatalysisDocument3 pagesTransition Metal Catalysisapi-250366166No ratings yet
- P-101 AlkynesDocument10 pagesP-101 AlkynesNISARG PATKARNo ratings yet
- PART TEST-1 (Ogranic Chemistry) PDFDocument6 pagesPART TEST-1 (Ogranic Chemistry) PDFKinshuk RastogiNo ratings yet
- Haloalkanes and Haloarenes-15 Mar 23Document5 pagesHaloalkanes and Haloarenes-15 Mar 23akshat.sh2021No ratings yet
- 8-Birch Reduction PDFDocument7 pages8-Birch Reduction PDFVENUGOPALARAO100% (1)
- LipidsDocument8 pagesLipidsBILL LLONARD RESURRECCIONNo ratings yet
- Alkanes, Alkenes, Alkunas and Cycloalkanes: Discussion GroupsDocument62 pagesAlkanes, Alkenes, Alkunas and Cycloalkanes: Discussion GroupsNing CahNo ratings yet
- Arihant Chemistry HandBookDocument14 pagesArihant Chemistry HandBookKannada SubjectNo ratings yet
- Non-Hydrocarbon - Esters: RCOOR Where R and R Represented The Same or Different Alkyl GroupsDocument2 pagesNon-Hydrocarbon - Esters: RCOOR Where R and R Represented The Same or Different Alkyl Groupscikgu ayuNo ratings yet
- Alcohol Phenol Ether and Carbonyl Compounds. Assignment Q. (Adv) .Document8 pagesAlcohol Phenol Ether and Carbonyl Compounds. Assignment Q. (Adv) .Anurag RamachandranNo ratings yet