You might also like
- Final Exam KeyDocument12 pagesFinal Exam KeykitthiNo ratings yet
- Organometallic CompoundsDocument66 pagesOrganometallic CompoundsJon Ho100% (1)
- Organometallic ChemistryDocument24 pagesOrganometallic ChemistryFatma TaherNo ratings yet
- 12 Chemistry Impq CH09 Coordination Compounds 01Document7 pages12 Chemistry Impq CH09 Coordination Compounds 01Sudarshan PandeyNo ratings yet
- Spectros PDFDocument28 pagesSpectros PDFbalajiNo ratings yet
- AlkeneDocument3 pagesAlkeneRichard MohammedNo ratings yet
- Experiment 4: Effect of Concentration and Temperature On Rate of Reaction (Dissappearing Cross)Document24 pagesExperiment 4: Effect of Concentration and Temperature On Rate of Reaction (Dissappearing Cross)Malini RajeshNo ratings yet
- CHEM F111: General Chemistry II-Semester Lecture 35 (12-04-2019Document20 pagesCHEM F111: General Chemistry II-Semester Lecture 35 (12-04-2019Rachit ShahNo ratings yet
- Determination of o of Chromium Using Tanabe-Sugano DiagramDocument2 pagesDetermination of o of Chromium Using Tanabe-Sugano DiagramDozdiNo ratings yet
- Automated Methods of AnalysisDocument9 pagesAutomated Methods of AnalysisPeerBuxNo ratings yet
- SN1 Vs SN2 ReactionsDocument23 pagesSN1 Vs SN2 Reactionssamnas100No ratings yet
- Isolobal AnalogyDocument11 pagesIsolobal AnalogyGA GANo ratings yet
- 1 IntroductoryDocument45 pages1 IntroductoryTuhin Sahu100% (1)
- Organometallic ChemistryDocument31 pagesOrganometallic ChemistrySadiaKhan100% (1)
- Aromaticity CompleteDocument104 pagesAromaticity Completewahidalwahdi100% (1)
- Experiment 1Document4 pagesExperiment 1JasmeetSinghNo ratings yet
- Hückel's MO Treatment of BenzeneDocument12 pagesHückel's MO Treatment of BenzeneRichard Allen0% (1)
- Potentiometric Methods - Q & ADocument34 pagesPotentiometric Methods - Q & AHoongNo ratings yet
- Acid Base Titration ExperimentDocument2 pagesAcid Base Titration ExperimentDark_KiroNo ratings yet
- Chemistry of Reactive Intermediate FinalDocument38 pagesChemistry of Reactive Intermediate FinalTefera100% (1)
- 01 1350977450 79497 PDFDocument83 pages01 1350977450 79497 PDFArya ChowdhuryNo ratings yet
- CAIE Chemistry A-Level: 24: ElectrochemistryDocument8 pagesCAIE Chemistry A-Level: 24: ElectrochemistryahumanbeinginearthNo ratings yet
- Department of Chemical EngineeringDocument12 pagesDepartment of Chemical EngineeringSheikh AliNo ratings yet
- Molecular Orbital Therory-Diatomic MoleculesDocument25 pagesMolecular Orbital Therory-Diatomic MoleculesDnyaneshwar ShindeNo ratings yet
- Lab ManualDocument19 pagesLab Manualanon_467104036No ratings yet
- Electron Delocalization and ResonanceDocument9 pagesElectron Delocalization and ResonanceMariana LizethNo ratings yet
- Bioinorganic HandoutDocument63 pagesBioinorganic HandoutAL__52No ratings yet
- Basic Principle and Applications of Paper ElectrophoresisDocument38 pagesBasic Principle and Applications of Paper ElectrophoresisSanty KoshyNo ratings yet
- Horseradish Peroxidase StudyDocument8 pagesHorseradish Peroxidase StudyRosyNotesNo ratings yet
- Ligand substitution mechanisms and rate laws in octahedral complexesDocument42 pagesLigand substitution mechanisms and rate laws in octahedral complexesLUIS CARLOS ROMERO ZAPATA100% (1)
- Transition Metals and Coordination ChemistryDocument80 pagesTransition Metals and Coordination ChemistryVincent Choo100% (1)
- Hypervalent Iodine: Dess-Martin Periodane: Selective Oxidation of Prim. Alcohols To Aldehydes, Sec. Alcohols To KetonesDocument15 pagesHypervalent Iodine: Dess-Martin Periodane: Selective Oxidation of Prim. Alcohols To Aldehydes, Sec. Alcohols To Ketonesevsgoud_goudNo ratings yet
- Cy 101 Uv-Vis and Ir NewDocument66 pagesCy 101 Uv-Vis and Ir NewSomesh MohapatraNo ratings yet
- Unit-3: Phase EquilibriaDocument94 pagesUnit-3: Phase EquilibriaNiboli K ZhimomiNo ratings yet
- Unit - 1 Lesson - 1Document271 pagesUnit - 1 Lesson - 1Rakesh SharmaNo ratings yet
- 12 Chemistry Ncert Ch09 Coordination Compounds Part 01 QuesDocument43 pages12 Chemistry Ncert Ch09 Coordination Compounds Part 01 Queshumayun khalidNo ratings yet
- ElecSpectra 2 UploadDocument25 pagesElecSpectra 2 UploadSarthak SinghNo ratings yet
- Assignment 8 - Solutions Chem1000ADocument4 pagesAssignment 8 - Solutions Chem1000AXdyne67% (3)
- Chemical Synthesis of Chalcones by Claisen-Schmidt Condensation Reaction and Its CharacterizationDocument5 pagesChemical Synthesis of Chalcones by Claisen-Schmidt Condensation Reaction and Its CharacterizationIJRASETPublicationsNo ratings yet
- Aromaticity PDFDocument9 pagesAromaticity PDFabyssabhi100% (2)
- Absorption Laws (Quantitative Analysis)Document15 pagesAbsorption Laws (Quantitative Analysis)Belay HaileNo ratings yet
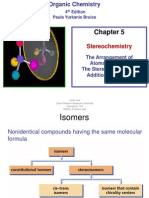
- Stereochemistry: 4 Edition Paula Yurkanis BruiceDocument43 pagesStereochemistry: 4 Edition Paula Yurkanis Bruicenrguerrerod100% (1)
- Bent RuleDocument24 pagesBent Rulesuka11blyatNo ratings yet
- 12 Chemistry Imp Isomerism in Coordination Compounds MixDocument8 pages12 Chemistry Imp Isomerism in Coordination Compounds MixMeha JabeenNo ratings yet
- Chapter 15 Parallels Between Main Group and Organometallic ChemistryDocument46 pagesChapter 15 Parallels Between Main Group and Organometallic ChemistryTarang BhatiNo ratings yet
- Importance of C-C Cross Coupling Reactions in Pharmaceutical ChemistryDocument67 pagesImportance of C-C Cross Coupling Reactions in Pharmaceutical ChemistryAnonymous vRpzQ2BLNo ratings yet
- Huckel Theory For Conjugated Systems: CH 105: Organic ChemistryDocument72 pagesHuckel Theory For Conjugated Systems: CH 105: Organic ChemistryRaunaq Bhirangi100% (1)
- Aromaticity Tutorial: Pi BondsDocument15 pagesAromaticity Tutorial: Pi BondsAlex-Mihai Ciubara100% (2)
- Lecture 22Document16 pagesLecture 22imania shaumiNo ratings yet
- Chapter 3 Coordination ChemistryDocument41 pagesChapter 3 Coordination Chemistrytarun ratnaNo ratings yet
- Lecture 6 Kinetic Isotope EffectDocument11 pagesLecture 6 Kinetic Isotope EffectcsnNo ratings yet
- Qualitative Treatment of Molecular Orbital TheoryDocument27 pagesQualitative Treatment of Molecular Orbital TheoryIfiok UsoroNo ratings yet
- Oxygen Containing Organic CompoundsDocument9 pagesOxygen Containing Organic CompoundsmNo ratings yet
- Classification of Organometallic CompoundsDocument28 pagesClassification of Organometallic CompoundsDingetegna GodanaNo ratings yet
- Potentiometry 2021Document47 pagesPotentiometry 2021Adina IqbalNo ratings yet
- F321 PeriodicityDocument3 pagesF321 PeriodicityDoc_CrocNo ratings yet
- Ideal and Non-Ideal SolutionsDocument3 pagesIdeal and Non-Ideal SolutionsJamshaid AliNo ratings yet
- Lecture 3: 2D NMR Spectroscopy 2D NMR Spectroscopy: Chem 135Document70 pagesLecture 3: 2D NMR Spectroscopy 2D NMR Spectroscopy: Chem 135ahmedkhidryagoubNo ratings yet
- Teachphil PDFDocument8 pagesTeachphil PDFahmedkhidryagoubNo ratings yet
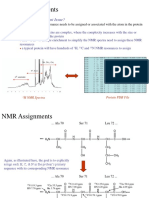
- NMR AssignmentsDocument87 pagesNMR AssignmentsahmedkhidryagoubNo ratings yet
- Gold Refining by Solvent Extraction-The Minataur™ ProcessDocument6 pagesGold Refining by Solvent Extraction-The Minataur™ ProcessahmedkhidryagoubNo ratings yet
- Sulphide Ore Test Results Reveal Potential for Agitation LeachingDocument50 pagesSulphide Ore Test Results Reveal Potential for Agitation LeachingahmedkhidryagoubNo ratings yet
- NANOEXC2004 Conference Abstracts Electronic Excitations Theory Modeling NanoscienceDocument52 pagesNANOEXC2004 Conference Abstracts Electronic Excitations Theory Modeling NanoscienceahmedkhidryagoubNo ratings yet
- PeptidNMR PDFDocument27 pagesPeptidNMR PDFahmedkhidryagoubNo ratings yet
- Teachphil PDFDocument8 pagesTeachphil PDFahmedkhidryagoubNo ratings yet
- Batforheapleaching Feb2013 C.zanbak EurominesDocument36 pagesBatforheapleaching Feb2013 C.zanbak EuromineslaughingalirezaNo ratings yet
- Wutrich LectureDocument33 pagesWutrich LectureMaksim MokeevNo ratings yet
- Gold DeportmentDocument17 pagesGold DeportmentahmedkhidryagoubNo ratings yet
- Analysis GoldDocument5 pagesAnalysis GoldahmedkhidryagoubNo ratings yet
- Nchem 1628Document13 pagesNchem 1628ahmedkhidryagoubNo ratings yet
- MikananobookDocument34 pagesMikananobookahmedkhidryagoubNo ratings yet
- DNA Holds the Secret to the Holy Grail of PhysicsDocument8 pagesDNA Holds the Secret to the Holy Grail of Physicsahmedkhidryagoub100% (1)
- Cisplatin FinalDocument27 pagesCisplatin FinalNadya HartasiwiNo ratings yet
- Ni Hms 91875Document31 pagesNi Hms 91875ahmedkhidryagoubNo ratings yet
- Thesis 1Document107 pagesThesis 1ahmedkhidryagoubNo ratings yet
- Darwin 200 BookletDocument23 pagesDarwin 200 BookletSuhana NanaNo ratings yet
- EvaluationDocument10 pagesEvaluationahmedkhidryagoubNo ratings yet
- The Process of Manufacture of Urea in A Naphtha Based PlantDocument4 pagesThe Process of Manufacture of Urea in A Naphtha Based PlantahmedkhidryagoubNo ratings yet
- Human EnthalpyDocument3 pagesHuman EnthalpyahmedkhidryagoubNo ratings yet
- Darfur The Road To PeaceDocument546 pagesDarfur The Road To Peaceahmedkhidryagoub100% (1)
- DNA Holds the Secret to the Holy Grail of PhysicsDocument8 pagesDNA Holds the Secret to the Holy Grail of Physicsahmedkhidryagoub100% (1)
- Anti-Israeili Terrorism in 2007 and Its Trends in 2008Document95 pagesAnti-Israeili Terrorism in 2007 and Its Trends in 2008ahmedkhidryagoub100% (1)
- Trends and Futures of Educatio1Document17 pagesTrends and Futures of Educatio1ahmedkhidryagoubNo ratings yet
- Human EnthalpyDocument3 pagesHuman EnthalpyahmedkhidryagoubNo ratings yet
- Constructivist I Constructivist Learning Environment Case Studies in Instructional Design IDocument16 pagesConstructivist I Constructivist Learning Environment Case Studies in Instructional Design IahmedkhidryagoubNo ratings yet
- Open Learning Meets The Business of Education: Trends in Distance LearningDocument5 pagesOpen Learning Meets The Business of Education: Trends in Distance LearningahmedkhidryagoubNo ratings yet
- Boiler Water TreatmentDocument50 pagesBoiler Water Treatmentak_thimiriNo ratings yet
- Form 1 Science - Unit 3.3: The Concept of DensityDocument1 pageForm 1 Science - Unit 3.3: The Concept of DensitySuhaila SaniNo ratings yet
- Mott - HyPulse Element FilterDocument12 pagesMott - HyPulse Element FilterCristhian CuevaNo ratings yet
- Scientific Paper Exp 5Document4 pagesScientific Paper Exp 5Brent TenorioNo ratings yet
- Pacop Red Pharmaceutical ChemistryDocument109 pagesPacop Red Pharmaceutical ChemistryAstherielle GalvezNo ratings yet
- 1.7 Batch Cell Culture 2Document36 pages1.7 Batch Cell Culture 2Astra BeckettNo ratings yet
- 45 - Miscible DisplacementDocument15 pages45 - Miscible Displacementrizal tri susilo67% (3)
- TheSouthernWarPoetryoftheCivilWar 10134200Document195 pagesTheSouthernWarPoetryoftheCivilWar 10134200Isaiah ThomasNo ratings yet
- Gravimetric Determination of Moisture CoDocument5 pagesGravimetric Determination of Moisture CoDEFIN BIMA REYNANDANo ratings yet
- 11.3 Relative Stability of Element Group 14Document13 pages11.3 Relative Stability of Element Group 14吴绍轩No ratings yet
- Johnson Matthey Syngas Methanol Plant Capacity FinalDocument14 pagesJohnson Matthey Syngas Methanol Plant Capacity FinalRaquel Siñani ChavezNo ratings yet
- PolarityDocument22 pagesPolarityEvangelene Esquillo SanaNo ratings yet
- A Study On The Compressive Strength and Water Absorption of Fired Soda Ash Infused Clay BricksDocument30 pagesA Study On The Compressive Strength and Water Absorption of Fired Soda Ash Infused Clay BricksNicole Andrei BaldozaNo ratings yet
- Pko Cno PDFDocument26 pagesPko Cno PDFmindcrNo ratings yet
- SDS Hydrochloric Acid InhibitorDocument4 pagesSDS Hydrochloric Acid InhibitorJahidul IslamNo ratings yet
- Test 2 CHM572 June 2024Document3 pagesTest 2 CHM572 June 2024NUR AINA SYAHMINA MOHD AMRANNo ratings yet
- Recipes For MilletsDocument20 pagesRecipes For MilletsRavi TejaNo ratings yet
- ISC ChemistryDocument8 pagesISC Chemistrysamrounder100% (3)
- ChemistryDocument22 pagesChemistrymacaronloverNo ratings yet
- Demulsifiers For Simulated Basrah Crude OilDocument107 pagesDemulsifiers For Simulated Basrah Crude OilTuan Yusoff100% (2)
- ISO 17025 NDT Lab Accreditation CertificateDocument3 pagesISO 17025 NDT Lab Accreditation Certificatekiki270977No ratings yet
- Lesson 2 Dna Structure and Dna ExtractionDocument8 pagesLesson 2 Dna Structure and Dna ExtractionGreatel Elijah TorregosaNo ratings yet
- Final Biodiesel ReportDocument60 pagesFinal Biodiesel ReportJohan LukitaNo ratings yet
- US4450090Document6 pagesUS4450090Abdulrahman HamdanNo ratings yet
- Orgo2 Lab Report1Document2 pagesOrgo2 Lab Report1api-382264943No ratings yet
- Hydrolysis Worksheet Chemistry Unit 4.13Document3 pagesHydrolysis Worksheet Chemistry Unit 4.13Gideon CavidaNo ratings yet
- Complexometrictitration-Ppt Part III A 2Document21 pagesComplexometrictitration-Ppt Part III A 2Anand NanavatyNo ratings yet
- Vidya Jyothi Chem Mock TestDocument6 pagesVidya Jyothi Chem Mock TestArko SarkarNo ratings yet
- PE PPBrochure - v1 - A4 - 110717Document8 pagesPE PPBrochure - v1 - A4 - 110717jamil ahmedNo ratings yet
- Chemistry Structure and Properties 2nd Edition Tro Test BankDocument24 pagesChemistry Structure and Properties 2nd Edition Tro Test Bankjenniferrichardsonjrwfpzsdim100% (29)