You might also like
- Structure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsFrom EverandStructure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsNo ratings yet
- Biochemistry Question Bank V 1.0e: Multiple Choice QuestionsDocument39 pagesBiochemistry Question Bank V 1.0e: Multiple Choice QuestionsArulkumar ManiNo ratings yet
- Crispr Cas 9Document3 pagesCrispr Cas 9E narender nayakNo ratings yet
- Mass SpectrosDocument47 pagesMass SpectrosEdward PittsNo ratings yet
- 2D NMRlatestDocument34 pages2D NMRlatestNandan ShindeNo ratings yet
- 1.5.0.potentials and EquilibriumDocument8 pages1.5.0.potentials and EquilibriumAnonymous G3DRjDMkNo ratings yet
- Contact AngleDocument7 pagesContact AngleOh Ha Ni OthmanNo ratings yet
- Biochemical TechniquesDocument4 pagesBiochemical TechniquesAyman ElsirNo ratings yet
- Csir Net Examination Life Sciences December 2012 PDFDocument77 pagesCsir Net Examination Life Sciences December 2012 PDFAbhay KumarNo ratings yet
- How Do You Calculate The Sensitivity in A Voltammetry AnalysisDocument11 pagesHow Do You Calculate The Sensitivity in A Voltammetry AnalysisElbahi DjaalabNo ratings yet
- Vibrational Analysis: Dr. Mazhar Amjad GilaniDocument23 pagesVibrational Analysis: Dr. Mazhar Amjad GilaniXarOonNo ratings yet
- SRM University Nano MaterialsDocument37 pagesSRM University Nano MaterialsVikram JainNo ratings yet
- Instrumental AnalysisDocument43 pagesInstrumental AnalysisGab Trinilla100% (1)
- Study Notes: The GC ColumnDocument16 pagesStudy Notes: The GC ColumnLaxmi Kant PrasadNo ratings yet
- The Spintronic Scanner For Cancer Detection Full ReportDocument29 pagesThe Spintronic Scanner For Cancer Detection Full ReportmolusssNo ratings yet
- Nano Material Synthesis Techniques Top-Down and Bottom-Up Nanofabrication, Synthesis ofDocument6 pagesNano Material Synthesis Techniques Top-Down and Bottom-Up Nanofabrication, Synthesis ofsanthoshNo ratings yet
- Biophysics Final QuestionsDocument4 pagesBiophysics Final Questionshadigy1001No ratings yet
- RFLPDocument1 pageRFLPachin47No ratings yet
- NepheloturbidometryDocument6 pagesNepheloturbidometryzaife khanNo ratings yet
- An Assignment On Ultraviolet and Visible SpectrometerDocument12 pagesAn Assignment On Ultraviolet and Visible SpectrometerSonnet100% (1)
- Quantum Espresso Inputfile ExplantionDocument31 pagesQuantum Espresso Inputfile ExplantionPHYSICS PROBLEMSNo ratings yet
- Pam BlosumDocument71 pagesPam Blosumrck46100% (1)
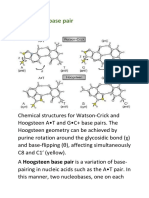
- Hoogsteen Base Pair PDFDocument7 pagesHoogsteen Base Pair PDFNikitaNo ratings yet
- Crispr Cas HajarDocument21 pagesCrispr Cas HajarHajira Fatima100% (1)
- Spectrophotometry. Principle and ApplicationsDocument11 pagesSpectrophotometry. Principle and Applicationsmdusman2010No ratings yet
- SPECTROSCOPY - Docx Senthil - Docx 1......Document17 pagesSPECTROSCOPY - Docx Senthil - Docx 1......Zaidh. fd100% (1)
- UV Vis InstrumentationDocument17 pagesUV Vis InstrumentationKD LoteyNo ratings yet
- Nucleic Acid Extraction MethodsDocument25 pagesNucleic Acid Extraction Methodsmasthan6yNo ratings yet
- Nuclear Magnetic ResonanceDocument23 pagesNuclear Magnetic ResonanceDavid OsasNo ratings yet
- Ion Trap Mass SpectrometryDocument8 pagesIon Trap Mass SpectrometryTommy BetyNo ratings yet
- 1 Dye-Sensitized Solar Cells: History, Components, Configuration, and Working PrincipleDocument16 pages1 Dye-Sensitized Solar Cells: History, Components, Configuration, and Working Principlefufumarifuu100% (1)
- Mass Spectrometry: Ev MVDocument12 pagesMass Spectrometry: Ev MVMuhammad Tariq RazaNo ratings yet
- Atomic Spectroscopy and Atomic Absorption SpectrosDocument80 pagesAtomic Spectroscopy and Atomic Absorption SpectrosMuhammad Mustafa IjazNo ratings yet
- MCQ On Probability & Random Signal Theory - Engineering MCQDocument9 pagesMCQ On Probability & Random Signal Theory - Engineering MCQPrajwal BirwadkarNo ratings yet
- Digoxin PPT Presentation FinalDocument16 pagesDigoxin PPT Presentation FinalMuhammad Rizky100% (1)
- Protein TargettingDocument11 pagesProtein TargettingPMIB Matrikulasi FKUI 2018/2019No ratings yet
- Smt. Kishoritai Bhoyar College of Pharmacy, New Kamptee.: BY Guided by Bhavik S.Kotak Dr. K.R.GuptaDocument33 pagesSmt. Kishoritai Bhoyar College of Pharmacy, New Kamptee.: BY Guided by Bhavik S.Kotak Dr. K.R.Guptadil_009100% (3)
- Chemistry PDFDocument78 pagesChemistry PDFNicholas SaahNo ratings yet
- Bragg'S Law, and Diffractometer: by Ayesha SiddiqaDocument11 pagesBragg'S Law, and Diffractometer: by Ayesha SiddiqaAyesha SiddiqaNo ratings yet
- Optical SpectrosDocument7 pagesOptical SpectrosgayaxniNo ratings yet
- Maldi TofDocument42 pagesMaldi TofMustafa AltındişNo ratings yet
- Slide Uv VisDocument54 pagesSlide Uv VisElka Sushea IINo ratings yet
- Purification of DNADocument14 pagesPurification of DNAalivetutorsNo ratings yet
- Rotational Raman Spectroscopy: The Polarizability of The Molecule Must Be AnisotropicDocument21 pagesRotational Raman Spectroscopy: The Polarizability of The Molecule Must Be AnisotropicKartik RanaNo ratings yet
- R Data Analysis Examples - Canonical Correlation AnalysisDocument7 pagesR Data Analysis Examples - Canonical Correlation AnalysisFernando AndradeNo ratings yet
- Nuclear Magnetic Resonance: Half-Integer Odd Odd or EvenDocument19 pagesNuclear Magnetic Resonance: Half-Integer Odd Odd or EvenRAJ VYASNo ratings yet
- PolarimetryDocument9 pagesPolarimetryAshaq HussainNo ratings yet
- High Performance Liquid ChromatographyDocument28 pagesHigh Performance Liquid ChromatographyNur Asiah0% (1)
- Kalyanasundaram, K. - Dye-Sensitized Solar Cells-Taylor & Francis (2010) PDFDocument355 pagesKalyanasundaram, K. - Dye-Sensitized Solar Cells-Taylor & Francis (2010) PDFJorge Reyes67% (3)
- NMR Lecture FinalDocument98 pagesNMR Lecture FinalXarOonNo ratings yet
- Environmental Chemistry PDFDocument3 pagesEnvironmental Chemistry PDFRijit ChakrabortyNo ratings yet
- Ap Chemistry: Kinetics Practice Problems: Rate of Reaction - (Clo (Clo (CL ) ) ) 3 2 T T TDocument13 pagesAp Chemistry: Kinetics Practice Problems: Rate of Reaction - (Clo (Clo (CL ) ) ) 3 2 T T TAbu Sufyan ButtNo ratings yet
- Unit 4 Chromatography-IIDocument18 pagesUnit 4 Chromatography-IIAli SheikhNo ratings yet
- CHAPTER-chemwiki Mossbauer SpectrosDocument8 pagesCHAPTER-chemwiki Mossbauer SpectrosAparna PrasadNo ratings yet
- 05Document43 pages05Sania ZahoorNo ratings yet
- Protein PredictionDocument100 pagesProtein Predictionvani_darlingNo ratings yet
- PH 107 Quantum Physics: Jagatap@phy - Iitb.ac - inDocument44 pagesPH 107 Quantum Physics: Jagatap@phy - Iitb.ac - inKasi Reddy Sreeman ReddyNo ratings yet
- Lecture 3 Significance of Biology in EngineeringDocument30 pagesLecture 3 Significance of Biology in EngineeringSaakshi PalNo ratings yet
- Ragone - Enunciados Problemas (Cap. 1 A 5)Document14 pagesRagone - Enunciados Problemas (Cap. 1 A 5)LucioNo ratings yet
- Reinforced Concrete Design-Krishnaraju PDFDocument318 pagesReinforced Concrete Design-Krishnaraju PDFJustin100% (4)
- Flexural Behaviour of RCC Beams: S TejaswiDocument5 pagesFlexural Behaviour of RCC Beams: S TejaswiJdjdjsjsNo ratings yet
- LATEST RWF-Prop-1-cast-vs-forge-modifiedDocument8 pagesLATEST RWF-Prop-1-cast-vs-forge-modifiedDevarshi GaurNo ratings yet
- Review PPT 2023Document7 pagesReview PPT 2023Cjs GamingNo ratings yet
- Handbook PolymeDocument9 pagesHandbook PolymeTrùm Dầu Mỏ BkNo ratings yet
- ASTM D 4014 3 BearingsDocument7 pagesASTM D 4014 3 Bearingssruthi raniNo ratings yet
- Failure of PigtailsDocument32 pagesFailure of Pigtailsbarry nancoo100% (1)
- SpringsDocument52 pagesSpringsrohan malikNo ratings yet
- Continuous Distillation Diagram ProcessDocument1 pageContinuous Distillation Diagram ProcessSebastianNo ratings yet
- Microsoft Word - B.sc. Physics SllybusDocument3 pagesMicrosoft Word - B.sc. Physics SllybusRahul YadavNo ratings yet
- Mesri 1997Document11 pagesMesri 1997RivaiNo ratings yet
- 2016 Practical Guide PDFDocument9 pages2016 Practical Guide PDFPromiseDoringtenNkosiNo ratings yet
- Chapter 2 Alloys - 2012 - Applied Welding EngineeringDocument5 pagesChapter 2 Alloys - 2012 - Applied Welding EngineeringJames LeonNo ratings yet
- Mechanics of MasonryDocument38 pagesMechanics of MasonryMyrto TsitsinakiNo ratings yet
- Selective LeachingDocument13 pagesSelective Leachingarslanjaved690100% (1)
- Separation and Purification of Organic Compounds-DistillationDocument17 pagesSeparation and Purification of Organic Compounds-DistillationMarie Maraniag100% (1)
- Computer Science ProjectDocument6 pagesComputer Science ProjectMohit KumarNo ratings yet
- 18 Japan2012 Milovan Peric VOFDocument39 pages18 Japan2012 Milovan Peric VOFAndreaNo ratings yet
- Fired Heaters and Boilers InspectionDocument31 pagesFired Heaters and Boilers Inspectionriysall100% (1)
- Chapter 4 MEMS Micro Sensors and ActuatorsDocument12 pagesChapter 4 MEMS Micro Sensors and ActuatorstcsNo ratings yet
- Determination of Solidus and Liquidus Alloys Temperatures 97831 r2Document1 pageDetermination of Solidus and Liquidus Alloys Temperatures 97831 r2safarNo ratings yet
- BARC Interview NotesDocument6 pagesBARC Interview NotesrajeshNo ratings yet
- Course:: Refrigeration and Air-Conditioning (ME 331)Document34 pagesCourse:: Refrigeration and Air-Conditioning (ME 331)Ch. Muhammad UsamaNo ratings yet
- K12 For Timber Designers' Manuel by MeDocument68 pagesK12 For Timber Designers' Manuel by MesajeeralaNo ratings yet
- Surface Characteristics ImplantsDocument83 pagesSurface Characteristics ImplantsdrpriyamNo ratings yet
- Plasticizers For CPE ElastomersDocument8 pagesPlasticizers For CPE Elastomersbatur42No ratings yet
- Effect of Shot Peening Operation On Gears PDFDocument9 pagesEffect of Shot Peening Operation On Gears PDFasdNo ratings yet
- Science 7 Weekly Home Learning PlanDocument6 pagesScience 7 Weekly Home Learning PlanMark PechoNo ratings yet
- AComplete Analytical Modelfor Square Diaphragm Capcitive Sensor With Clamped EdgesDocument5 pagesAComplete Analytical Modelfor Square Diaphragm Capcitive Sensor With Clamped EdgesJonas HilarioNo ratings yet