You might also like
- Relajantes Del Músculo EsqueléticoDocument51 pagesRelajantes Del Músculo EsqueléticoLaura BulaNo ratings yet
- Anestésicos LocalesDocument30 pagesAnestésicos LocalesDany Ramos López100% (1)
- Síntesis de ProstaglandinasDocument8 pagesSíntesis de ProstaglandinasLucero Guadalupe Flores DiazNo ratings yet
- Mycobacterium LepraeDocument42 pagesMycobacterium LepraedangrangerNo ratings yet
- Procesamiento SensitivoDocument56 pagesProcesamiento SensitivoMario Catalán Faúndez100% (1)
- Tipos de AntineoplasicosDocument5 pagesTipos de Antineoplasicosgustavoconde1983100% (1)
- Toxicidad de medicamentos en oncologíaDocument55 pagesToxicidad de medicamentos en oncologíaGuadalupeFigueroaLópez100% (2)
- Agonistas y Antagonistas ColinérgicosDocument20 pagesAgonistas y Antagonistas ColinérgicosKevin victor Carbajal CarhuaricraNo ratings yet
- Receptores ColinergicosDocument17 pagesReceptores ColinergicosJosé A. Beltran100% (1)
- Xenobioticos 2017Document40 pagesXenobioticos 2017Syovis HopeNo ratings yet
- Fármacos AntihiperlipidémicosDocument28 pagesFármacos Antihiperlipidémicosleentao100% (1)
- Mycobacterium 2Document43 pagesMycobacterium 2Juan Wilfredo Hernandez DiazNo ratings yet
- BETALACTÁMICOSDocument15 pagesBETALACTÁMICOSKarol Stephany Cifuentes SanchezNo ratings yet
- AntiparkinsonianosDocument18 pagesAntiparkinsonianosGustavo Tovar50% (2)
- IbuprofenoDocument6 pagesIbuprofenoMarcelo Muñoz VegaNo ratings yet
- ERRORES INNATOS DEL METABOLISMO DE LOS LDocument42 pagesERRORES INNATOS DEL METABOLISMO DE LOS Lmarie1690No ratings yet
- Polimiositis: causas, síntomas y tratamientoDocument21 pagesPolimiositis: causas, síntomas y tratamientoGerlit Camarena100% (1)
- Sindrome de Cordon AnteriorDocument4 pagesSindrome de Cordon Anteriorpatymend23No ratings yet
- Naftaleno TrabajoDocument16 pagesNaftaleno TrabajoAldair MedinaNo ratings yet
- AntihistaminicosDocument6 pagesAntihistaminicosAnthony VasconezNo ratings yet
- La GlucolisisDocument36 pagesLa GlucolisisMaria Angelica Santillan CarrascoNo ratings yet
- PentobarbitalDocument8 pagesPentobarbitalJordancinnNo ratings yet
- Diabetes Mellitus Tipo 1 Final 170511032148 PDFDocument42 pagesDiabetes Mellitus Tipo 1 Final 170511032148 PDFSus ArNo ratings yet
- FrancisellaDocument10 pagesFrancisellaJusal Palomino GalindoNo ratings yet
- Padrinos Magicos - Bloque 2Document8 pagesPadrinos Magicos - Bloque 2Paola Acosta100% (1)
- Enfermedades Infecciosas Del Sistema Nervioso CentralDocument30 pagesEnfermedades Infecciosas Del Sistema Nervioso Centralgiovanni cordovaNo ratings yet
- ColeraDocument50 pagesColeraJhon Alexander Suescún SepúlvedaNo ratings yet
- Inmunidad frente a parásitos: respuesta adaptativaDocument3 pagesInmunidad frente a parásitos: respuesta adaptativaAna PaulaNo ratings yet
- Bordetella PertussisDocument19 pagesBordetella PertussisZulema Garcia100% (1)
- Lipidos No SaponificablesDocument35 pagesLipidos No Saponificablesapi-3711538100% (1)
- Antihistaminicos Tipo h2 Ii3Document35 pagesAntihistaminicos Tipo h2 Ii3freddy_vasquez_15No ratings yet
- Radicales Libres y Diabetes MelllitusDocument14 pagesRadicales Libres y Diabetes Melllitusholimmm100% (1)
- Caso Clinico SepsisDocument5 pagesCaso Clinico SepsisAndres ViloriaNo ratings yet
- 2da Clase - Penicilina y Betalactamicos 2023Document69 pages2da Clase - Penicilina y Betalactamicos 2023Noriena MartinezNo ratings yet
- Linfocitos TDocument8 pagesLinfocitos TCristian Ignacio JaqueNo ratings yet
- Enfermedades PrionicasDocument13 pagesEnfermedades PrionicasTona PGNo ratings yet
- Reporte de AntiulcerososDocument8 pagesReporte de AntiulcerososА̵̨̨͙̬̳̦̟̼͇͓̐̈́͝͝р̵̛̮̈́̉͐͑͗̈́̓͗͐̈́͝э̴̭̥̝̲̐͋̆̆̋̆̀н̴̡̲̗̳̮̯̫̯̱́͂̋̈̽̈́̈́͊̈́͂̀̊̇͂̿̍͝͠ АрэнNo ratings yet
- Ramírez Morales Tarea BTDocument6 pagesRamírez Morales Tarea BTErick RMNo ratings yet
- Antiagregantes PlaquetariosDocument8 pagesAntiagregantes PlaquetariosYesZii Frezzitha PalaciosNo ratings yet
- Antidiabéticos Orales DiapositivasDocument42 pagesAntidiabéticos Orales DiapositivasAnyeli AnyeliNo ratings yet
- Tosferina 1Document4 pagesTosferina 1jasj_20100% (11)
- GLUCOSADocument14 pagesGLUCOSAFernanda VilchesNo ratings yet
- ANTICONVULSIVANTESDocument10 pagesANTICONVULSIVANTESVanesa ChávezNo ratings yet
- Adenitis PDFDocument7 pagesAdenitis PDFDavid MedranoNo ratings yet
- Tranquilizantes MayoresDocument65 pagesTranquilizantes MayoresMonse RodriguezNo ratings yet
- Farmaco Sistema Nervioso Central FDocument46 pagesFarmaco Sistema Nervioso Central FDayanaNo ratings yet
- Transportadores de GlucosaDocument10 pagesTransportadores de GlucosaJose AlvarezNo ratings yet
- Farmacologia Del Sistema RespirtorioDocument55 pagesFarmacologia Del Sistema RespirtorioLinda Santiago MarianoNo ratings yet
- 5 FluoroquinolonasDocument44 pages5 FluoroquinolonasCátedra de Farmacología de la Escuela de Medicina de La UNIVERSIDAD DEL ZULIANo ratings yet
- Virus 130706131217 Phpapp01Document35 pagesVirus 130706131217 Phpapp01jorgecremadesNo ratings yet
- Inhibidor EnzimáticoDocument18 pagesInhibidor EnzimáticoAdri CruzNo ratings yet
- Intoxicación Por Metanol PDFDocument4 pagesIntoxicación Por Metanol PDFBibiana VarelaNo ratings yet
- ColinergicosDocument26 pagesColinergicosJose Milton Morales GonzalesNo ratings yet
- HipersensibilidadDocument6 pagesHipersensibilidadDarwin Berrios AnacletoNo ratings yet
- DigoxinaDocument7 pagesDigoxinaclau1255No ratings yet
- HipolipemiantesDocument21 pagesHipolipemiantesKelly SVNo ratings yet
- BLOQUEADORES DopaminergicosDocument28 pagesBLOQUEADORES DopaminergicosJoseph Angel VINo ratings yet
- 6 DemenciaDocument35 pages6 DemenciafabioNo ratings yet
- Enfermedad de AlzheimerDocument50 pagesEnfermedad de AlzheimerAgustina IndovinaNo ratings yet
- Función renal y sus evaluacionesDocument37 pagesFunción renal y sus evaluacionesDenia EstradaNo ratings yet
- Homunculos CorticalesDocument2 pagesHomunculos CorticalesVanesa Camila Málaga SalcedoNo ratings yet
- Aparato Genital FemeninoDocument74 pagesAparato Genital FemeninoMarisabel LizanoNo ratings yet
- Tema 5 Tracto UvealDocument42 pagesTema 5 Tracto UvealJhoanna ParedesNo ratings yet
- Fisiopatologia Del AncianoDocument24 pagesFisiopatologia Del Ancianoluis andres trejo castellanosNo ratings yet
- RESIDENTESSSSSSSSSSDocument38 pagesRESIDENTESSSSSSSSSSDenise RomeroNo ratings yet
- Trastornos Del Ritmo RespiratorioDocument17 pagesTrastornos Del Ritmo RespiratorioCarLiita CruzNo ratings yet
- Histología: Tejido EpitelialDocument3 pagesHistología: Tejido EpitelialLizeth Olivares VelasquezNo ratings yet
- Anatomia Quirurgica Del Higado Postgrado Cirugia General HLV DR David YèpezDocument49 pagesAnatomia Quirurgica Del Higado Postgrado Cirugia General HLV DR David YèpezDavid Yépez YépezNo ratings yet
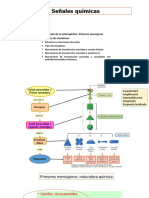
- Tema 6 Señales QuímicasDocument20 pagesTema 6 Señales QuímicasMar Ruiz Sainz-PardoNo ratings yet
- Muestras de Sangre AVESDocument8 pagesMuestras de Sangre AVESJuan NfrNo ratings yet
- Taller de Repaso Lípidos y Ácidos NucleicosDocument3 pagesTaller de Repaso Lípidos y Ácidos NucleicosVIKY VANESSA NAVIA MUñOZNo ratings yet
- Glandulas SalivalesDocument13 pagesGlandulas SalivalesChristopher Gallo TapiaNo ratings yet
- Informe de Práctica Sistema Digestivo IDocument5 pagesInforme de Práctica Sistema Digestivo IYoselyn MorochoNo ratings yet
- La LechugaDocument8 pagesLa LechugaElvis VillanuevaNo ratings yet
- EndocrinoDocument10 pagesEndocrinoKaren MendozaNo ratings yet
- Trabajo de Parto El Feto y El CanalDocument31 pagesTrabajo de Parto El Feto y El CanalMaria J. RendonNo ratings yet
- Ciclo Del SueñoDocument1 pageCiclo Del SueñoNicole RodriguezNo ratings yet
- Endocitosis y ExocitosisDocument5 pagesEndocitosis y ExocitosiscindyNo ratings yet
- Efecto de Los Radicales Libres en Pacientes DiabeticosDocument3 pagesEfecto de Los Radicales Libres en Pacientes DiabeticosCinthia Joallis HernandezNo ratings yet
- Mapa Conceptual NucleoDocument2 pagesMapa Conceptual NucleoNorma Yesica Garay Mejia86% (14)
- Corre Despacio para Correr Más Rápido - FRDocument6 pagesCorre Despacio para Correr Más Rápido - FREnzo100% (3)
- Solucion A Glande Inflamado o DebajoDocument2 pagesSolucion A Glande Inflamado o DebajoAgustinZavaletaNo ratings yet
- Silabo Estructura y Funcion Ii IntegradoDocument12 pagesSilabo Estructura y Funcion Ii IntegradoAna Luisa Guevara PrietoNo ratings yet
- Padron de Daños No Transmisibles (2019)Document47 pagesPadron de Daños No Transmisibles (2019)Anthony Enrique Arrieta GarayarNo ratings yet
- Fotosíntesis procesoDocument12 pagesFotosíntesis procesoDaniza Hidalgo PintoNo ratings yet
- CoartacionDocument17 pagesCoartacionLos Chaveztubies100% (1)
- ContagiousDocument452 pagesContagiousMaría Angela UbillúsNo ratings yet
- Silabo de Quimica Organica IiDocument8 pagesSilabo de Quimica Organica IiRocio Del Pilar QoNo ratings yet
- Reino Metazoos o Reino AnimalDocument28 pagesReino Metazoos o Reino AnimalTERESA LLORENTE DEL TORONo ratings yet
- Angiografía FluoresceìnicaDocument55 pagesAngiografía FluoresceìnicaSilvio Hidalgo100% (2)