You might also like
- GPC Hepatitis CDocument60 pagesGPC Hepatitis CEduardNo ratings yet
- FormatoLlenadoViaInternet NDocument2 pagesFormatoLlenadoViaInternet Nsee_userNo ratings yet
- NDocument47 pagesNsee_userNo ratings yet
- Enfermedad de Fabry (EyR) Copia 2Document71 pagesEnfermedad de Fabry (EyR) Copia 2see_userNo ratings yet
- NOM Tuberculosis 2013Document15 pagesNOM Tuberculosis 2013see_userNo ratings yet
- Raquitismo Carencial (EyR)Document47 pagesRaquitismo Carencial (EyR)see_userNo ratings yet
- Convocatoria Servicio Social 2018Document10 pagesConvocatoria Servicio Social 2018see_userNo ratings yet
- Diagnostico y Tratamiento de Hipotiroidismo Primario y Subclinico en El AdultoDocument46 pagesDiagnostico y Tratamiento de Hipotiroidismo Primario y Subclinico en El AdultoVirginia Alejandra GutierrezNo ratings yet
- Convocatoria Servicio Social 2018Document10 pagesConvocatoria Servicio Social 2018see_userNo ratings yet
- Pediatricos Con FenilcetonuriaDocument52 pagesPediatricos Con FenilcetonuriaYuli AguilarNo ratings yet
- GER DislipidemiaDocument68 pagesGER DislipidemiaMARIANA ESMERALDA LOPEZNo ratings yet
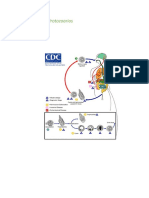
- Parásitos-Ciclos de VidaDocument24 pagesParásitos-Ciclos de Vidasee_userNo ratings yet
- Introducción A Dropbox PDFDocument10 pagesIntroducción A Dropbox PDFdavidNo ratings yet
- OftalmologíaDocument122 pagesOftalmologíasee_userNo ratings yet
- BuhoDocument54 pagesBuhosee_userNo ratings yet
- VulvovaginitisDocument14 pagesVulvovaginitisRodrigo Borja0% (1)
- Unidad Asist Circ Comp PerifericaDocument1 pageUnidad Asist Circ Comp PerifericaJoel GomezNo ratings yet
- Introduccion A La NutricionDocument26 pagesIntroduccion A La NutricionLorena Goetschel100% (3)
- EduSalud-40Document3 pagesEduSalud-40SHEYLA EVELYN DELGADO GAMERONo ratings yet
- HomotoxicologíaDocument5 pagesHomotoxicologíaRafa RodriguezNo ratings yet
- MAIS Adulto MayorDocument17 pagesMAIS Adulto MayorGrupo9PeralvilloNo ratings yet
- Fase 4 Grupo 151005 - 13Document28 pagesFase 4 Grupo 151005 - 13David GoyesNo ratings yet
- Factores de Riesgo DengueDocument1 pageFactores de Riesgo DengueAna ReyesNo ratings yet
- 1028787388-Danna ContrerasDocument44 pages1028787388-Danna ContrerasGabriel Fernando Baquero RomeroNo ratings yet
- Pancreatitis CrónicaDocument10 pagesPancreatitis CrónicaAlex AlvarezNo ratings yet
- Hoja Informativa 1Document2 pagesHoja Informativa 1emas04No ratings yet
- Programa Pcii Semestre 22-2Document29 pagesPrograma Pcii Semestre 22-2Ale Ávalos LeónNo ratings yet
- Infecciones de Transmision SexualDocument4 pagesInfecciones de Transmision SexualChinoMarrufoNo ratings yet
- Andrea Olguin, Reunion Noviembre PDFDocument24 pagesAndrea Olguin, Reunion Noviembre PDFCami ArosNo ratings yet
- Análisis corporal completoDocument2 pagesAnálisis corporal completoluzaleydaNo ratings yet
- SA-S2G1-V2Guia Morbilidad Oral PDFDocument78 pagesSA-S2G1-V2Guia Morbilidad Oral PDFYurani Zapata HernandezNo ratings yet
- Sílabo Clínica y Terapéutica en Medicina I 2021-I - 20210323215229Document24 pagesSílabo Clínica y Terapéutica en Medicina I 2021-I - 20210323215229adrianoNo ratings yet
- PROCEDIMIENTO DE TRABAJO SEGURO YESO Y PINTURA - Rev 01Document9 pagesPROCEDIMIENTO DE TRABAJO SEGURO YESO Y PINTURA - Rev 01Harold ValdebenitoNo ratings yet
- Ácido Hialurónico - Restylane FICHA TÉCNICADocument2 pagesÁcido Hialurónico - Restylane FICHA TÉCNICAjules39100% (1)
- Extractos Consenso Global Sobre Queratocono y Enfermedades Ectasicas Corneales Abril 2015Document7 pagesExtractos Consenso Global Sobre Queratocono y Enfermedades Ectasicas Corneales Abril 2015Valentina ÁlvarezNo ratings yet
- Preguntas Área RuralDocument11 pagesPreguntas Área RuralMaribel Yugar TarquiNo ratings yet
- Terapia de OrejaDocument14 pagesTerapia de OrejaJorgec08100% (1)
- Silabo de Nutricion - 2019Document22 pagesSilabo de Nutricion - 2019Mariana PitaNo ratings yet
- PIELES SecasDocument2 pagesPIELES SecasSABRINA NOEMI RODRIGUEZNo ratings yet
- Actividad Evaluativa Eje 4 ECONOMIA EN SALUDDocument20 pagesActividad Evaluativa Eje 4 ECONOMIA EN SALUDPatricia VianaNo ratings yet
- Hospital Amazónico: Sustento de Tomografia Cerebral ComputarizadaDocument7 pagesHospital Amazónico: Sustento de Tomografia Cerebral Computarizadaanon_389620856No ratings yet
- Actividad 2 Modulo 2Document3 pagesActividad 2 Modulo 2Brenda Javier Moreno100% (1)
- Guía de Prevención y Control de La Trasmisión Del VIH Sífilis Congénita y de Atención Integral Niños Con VIH SIDA PDFDocument154 pagesGuía de Prevención y Control de La Trasmisión Del VIH Sífilis Congénita y de Atención Integral Niños Con VIH SIDA PDFFrancisco AcostaNo ratings yet
- ApositosDocument3 pagesApositosSydney LópezNo ratings yet
- PROGRAMA DE AREAS Hospital PsiquiátricoDocument7 pagesPROGRAMA DE AREAS Hospital PsiquiátricoMoigually33% (6)