You might also like
- Patrick4e Ch21 PKDocument46 pagesPatrick4e Ch21 PKnosaybaNo ratings yet
- Patrick4e Ch21 PKDocument46 pagesPatrick4e Ch21 PKnosaybaNo ratings yet
- 2006-CHM6108 - L5L6 SlidesDocument52 pages2006-CHM6108 - L5L6 Slidesaidar.seralinNo ratings yet
- Metabolic Pathways: Glycolysis Pentose Phosphate PathwayDocument1 pageMetabolic Pathways: Glycolysis Pentose Phosphate PathwayLucasLeãoNascimentoNo ratings yet
- 2 Target Obat - 2022Document95 pages2 Target Obat - 2022Yeremi EgaNo ratings yet
- Osteoprorosis Therapy Management, Beyond Safety and Efficacy (Once Yearly Treatment)Document29 pagesOsteoprorosis Therapy Management, Beyond Safety and Efficacy (Once Yearly Treatment)ian ismeNo ratings yet
- 14glucose6 PpsDocument2 pages14glucose6 PpsSanjeefKumrIINo ratings yet
- Formation of Glucose 6-PhosphateDocument2 pagesFormation of Glucose 6-PhosphateSanjeefKumrIINo ratings yet
- Biosynthesis of Fatty Acids #1Document68 pagesBiosynthesis of Fatty Acids #1QOTRUNNADA AIDANo ratings yet
- Regulatory Enzyme MechanismsDocument15 pagesRegulatory Enzyme MechanismsoczhinviaNo ratings yet
- What Is Organic Chemistry?Document72 pagesWhat Is Organic Chemistry?gs7514424No ratings yet
- Severe Big Toe Pain May Indicate GoutDocument44 pagesSevere Big Toe Pain May Indicate GoutSAIMA PARVEENNo ratings yet
- Transcription Transcription - Translation Information Flow in Biological Systems - DNA Replication PDFDocument90 pagesTranscription Transcription - Translation Information Flow in Biological Systems - DNA Replication PDFAveen ShabanNo ratings yet
- Chapter 4 ContiuedDocument24 pagesChapter 4 ContiuedumarNo ratings yet
- Nucleic Acid Structure & DNA ReplicationDocument51 pagesNucleic Acid Structure & DNA ReplicationJMCDUFFIENo ratings yet
- Energy Production: Hapter 3Document18 pagesEnergy Production: Hapter 3Supriya A SNo ratings yet
- DNA, RNA & PROTEINS SYNTHESISDocument60 pagesDNA, RNA & PROTEINS SYNTHESISjaya1129No ratings yet
- Biosynthetic Pathway of PenicillinsDocument1 pageBiosynthetic Pathway of PenicillinskuganatsukiNo ratings yet
- Purine Metabolism II: Osama YousefDocument14 pagesPurine Metabolism II: Osama YousefWan NurfazliyanaNo ratings yet
- Lec 5Document20 pagesLec 5Sreemanti DeyNo ratings yet
- Glycolysis: The Energy-Extracting PathwayDocument48 pagesGlycolysis: The Energy-Extracting PathwayTeodora MunteanuNo ratings yet
- Curs 12 GlycogenDocument37 pagesCurs 12 GlycogenStanescuRozicaNo ratings yet
- 2006-CHM6108 - L5L6 HandoutDocument9 pages2006-CHM6108 - L5L6 Handoutaidar.seralinNo ratings yet
- 5.36 Biochemistry Laboratory: Mit OpencoursewareDocument11 pages5.36 Biochemistry Laboratory: Mit OpencoursewareNeenu RajputNo ratings yet
- Nucleotide Metabolism - Part 1: Purine Biosynthesis PathwayDocument49 pagesNucleotide Metabolism - Part 1: Purine Biosynthesis PathwayMohammed Ismail HegazyNo ratings yet
- Lipids Chapter - Terpenes and Terpenoids Natural ProductsDocument21 pagesLipids Chapter - Terpenes and Terpenoids Natural ProductsDaniel TranNo ratings yet
- The β - D Glucose Scaffold as a β- Turn Mimetic: Saidulu DaraDocument19 pagesThe β - D Glucose Scaffold as a β- Turn Mimetic: Saidulu Daraglreddy09No ratings yet
- Presentazione Tesi Matteo CerrinaDocument82 pagesPresentazione Tesi Matteo CerrinaMatteo CerrinaNo ratings yet
- Kami Export - 6 Biological MoleculesDocument5 pagesKami Export - 6 Biological MoleculesNicholas Crowell100% (1)
- Chemistry NoteDocument27 pagesChemistry NoteUmar M abubakarNo ratings yet
- Antibiotics Deraya 2Document33 pagesAntibiotics Deraya 2ahmed montaserNo ratings yet
- Lipids 2Document6 pagesLipids 2cumbredinNo ratings yet
- Honey Health Benefits and Uses in Medicine: Hana Scepankova, Jorge A. Saraiva, and Letícia M. EstevinhoDocument14 pagesHoney Health Benefits and Uses in Medicine: Hana Scepankova, Jorge A. Saraiva, and Letícia M. EstevinhoAndrés Felipe Zapata MurielNo ratings yet
- Further reading provides more contextDocument1 pageFurther reading provides more contextAmir ali WalizadehNo ratings yet
- 02 - Haze FormationDocument22 pages02 - Haze FormationGiorgi GhambashidzeNo ratings yet
- DNA PPSXDocument19 pagesDNA PPSXTintin Brusola SalenNo ratings yet
- K7 - Metabolisme NukleotidaDocument33 pagesK7 - Metabolisme NukleotidaAditya MuchayatsyahNo ratings yet
- K7 - Metabolisme NukleotidaDocument32 pagesK7 - Metabolisme NukleotidaTiara YosephaNo ratings yet
- Biopolymers LectureDocument23 pagesBiopolymers LectureNneka Okafor-EzeaniNo ratings yet
- Biological Macromolecules: Proteins, Nucleic Acids and LipidsDocument8 pagesBiological Macromolecules: Proteins, Nucleic Acids and LipidsRintuNo ratings yet
- Bioorganic Chemistry and Biochemistry CHM3218 Summer C 2008: Class WebsiteDocument45 pagesBioorganic Chemistry and Biochemistry CHM3218 Summer C 2008: Class WebsiteMadhu MattaNo ratings yet
- DNA ReplicationDocument20 pagesDNA ReplicationFrie An PanteNo ratings yet
- Available compound as a starting material for organic synthesisDocument10 pagesAvailable compound as a starting material for organic synthesisAfifaNo ratings yet
- 10 - Beer Stabilization Technology-Clearly A Matter of ChoiceDocument42 pages10 - Beer Stabilization Technology-Clearly A Matter of ChoiceGiorgi GhambashidzeNo ratings yet
- Mulungushi University Biochemistry Lecture on Nucleotide MetabolismDocument60 pagesMulungushi University Biochemistry Lecture on Nucleotide MetabolismEmmanuel ChendaNo ratings yet
- Tetrodotoxin: TTX: BackgroundDocument12 pagesTetrodotoxin: TTX: BackgroundMarrauNo ratings yet
- Prodrug (D.ashowq)Document4 pagesProdrug (D.ashowq)علي الطياريNo ratings yet
- Nucleotide Metabolism - Part 1 (Purine Biosynthesis)Document123 pagesNucleotide Metabolism - Part 1 (Purine Biosynthesis)Rohit VinayNo ratings yet
- Biosynthesis of Natural Products Derived From Shikimic Acid 4.1. Phenyl-Propanoid Natural Products (C - C)Document22 pagesBiosynthesis of Natural Products Derived From Shikimic Acid 4.1. Phenyl-Propanoid Natural Products (C - C)Preeti YadavNo ratings yet
- 9 GlycogenDocument36 pages9 GlycogenSneha Sagar SharmaNo ratings yet
- Alkaloid S1Document142 pagesAlkaloid S1Faisal MNo ratings yet
- Nucleic Acids: (Deoxyribonucleic Acids) (Ribonucleic Acids)Document45 pagesNucleic Acids: (Deoxyribonucleic Acids) (Ribonucleic Acids)Darshan TrivediNo ratings yet
- Nucleotides and Nucleic Acids: CH HO Base P O O O O CH Base P O O O O CH BaseDocument12 pagesNucleotides and Nucleic Acids: CH HO Base P O O O O CH Base P O O O O CH Basebiagio castronovoNo ratings yet
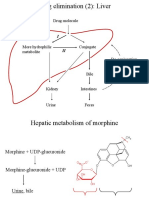
- Drug elimination (2): Liver metabolismDocument26 pagesDrug elimination (2): Liver metabolismSylvia AngelinaNo ratings yet
- Answers ch10Document6 pagesAnswers ch10김아진No ratings yet
- Pathways of Glucose Metabolism: Blood Glucose Rna & DnaDocument17 pagesPathways of Glucose Metabolism: Blood Glucose Rna & DnaAhmedNo ratings yet
- Pathways of Glucose Metabolism: Blood Glucose Rna & DnaDocument17 pagesPathways of Glucose Metabolism: Blood Glucose Rna & DnaReem AkraNo ratings yet
- Lec14 PDFDocument11 pagesLec14 PDFAishwarya MankaniNo ratings yet
- Seiple Oct 06Document13 pagesSeiple Oct 06Sơn Nguyễn KimNo ratings yet
- Learner Enrollment and Survey Form: Grade Level and School InformationDocument2 pagesLearner Enrollment and Survey Form: Grade Level and School InformationThe Great Papyrus90% (73)
- September: Sunday Monday Tuesday Wednesday Thursday Friday SaturdayDocument1 pageSeptember: Sunday Monday Tuesday Wednesday Thursday Friday SaturdaymaimaiNo ratings yet
- DRUG STABILITY TESTING PRINCIPLESDocument86 pagesDRUG STABILITY TESTING PRINCIPLESmaimaiNo ratings yet
- Milliequivalents, Millimoles, and Milliosmoles ExplainedDocument17 pagesMilliequivalents, Millimoles, and Milliosmoles ExplainedMuhammad SafdarNo ratings yet
- Exercise 23 - Sulfur OintmentDocument4 pagesExercise 23 - Sulfur OintmentmaimaiNo ratings yet
- Basic ConceptsDocument5 pagesBasic ConceptsmaimaiNo ratings yet
- IV Fluid Prescription PointsDocument1 pageIV Fluid Prescription PointsmaimaiNo ratings yet
- BP DeterminationDocument1 pageBP Determinationmaimai100% (1)
- Isoniazid Chemical ReactionsDocument1 pageIsoniazid Chemical ReactionsmaimaiNo ratings yet
- Veneration Without Understanding2Document10 pagesVeneration Without Understanding2Katrina SantosNo ratings yet
- BP DeterminationDocument1 pageBP Determinationmaimai100% (1)
- Assay Ferrous Sulfate TabletsDocument3 pagesAssay Ferrous Sulfate Tabletsmaimai67% (3)
- Veneration Without Understanding2Document10 pagesVeneration Without Understanding2Katrina SantosNo ratings yet
- Accts Comm First Sem AY '16-'17 ReportDocument9 pagesAccts Comm First Sem AY '16-'17 ReportmaimaiNo ratings yet
- Assay of Cupric SulfateDocument3 pagesAssay of Cupric Sulfatemaimai0% (2)
- Assay - Alumina and Magnesia Oral SuspensionDocument3 pagesAssay - Alumina and Magnesia Oral SuspensionmaimaiNo ratings yet
- The Partition of AfricaDocument6 pagesThe Partition of AfricamaimaiNo ratings yet
- UtangDocument11 pagesUtangmaimaiNo ratings yet
- It Imparts A Reddish ColorDocument3 pagesIt Imparts A Reddish Colormaimai100% (1)
- Assay of Lactic AcidDocument2 pagesAssay of Lactic Acidmaimai100% (1)
- Genealogical Tests Using DNADocument42 pagesGenealogical Tests Using DNAmaimaiNo ratings yet
- Assay of Cupric SulfateDocument3 pagesAssay of Cupric Sulfatemaimai0% (2)
- Assay Ferrous Sulfate TabletsDocument3 pagesAssay Ferrous Sulfate Tabletsmaimai67% (3)
- ДокументDocument72 pagesДокументDianna ArzumanovaNo ratings yet
- Novartis Case BGSDocument13 pagesNovartis Case BGSManoj Kumar SNo ratings yet
- Novartis Annual Report 2016Document274 pagesNovartis Annual Report 2016Kevin HaikalNo ratings yet
- Embolismo Pulmonar NejmDocument3 pagesEmbolismo Pulmonar NejmSaidNo ratings yet
- Index: Sr. No. Particulars Page NoDocument16 pagesIndex: Sr. No. Particulars Page NoNominee PareekNo ratings yet
- Jie Jack Li - Medicinal Chemistry For Practitioners-John Wiley & Sons Inc (2021)Document401 pagesJie Jack Li - Medicinal Chemistry For Practitioners-John Wiley & Sons Inc (2021)Rotoplastic de Cuauhtémoc S.A. de C.V.100% (1)
- CDER Fast Track Products ApprovedDocument5 pagesCDER Fast Track Products ApproveddianNo ratings yet
- PS31 - CML Booklet - 2019Document64 pagesPS31 - CML Booklet - 2019ameliaNo ratings yet
- Business Ethics of NovartisDocument20 pagesBusiness Ethics of NovartisSujata KumarNo ratings yet
- Case Study - Pharma IndustryDocument2 pagesCase Study - Pharma IndustryMazbahul IslamNo ratings yet
- Volume 2, Issue 2Document164 pagesVolume 2, Issue 2Asfandyar SheikhNo ratings yet
- What are not inventions under Indian Patent LawDocument29 pagesWhat are not inventions under Indian Patent LawNasif MustahidNo ratings yet
- IMN1207 McGillActiveProtocolsDocument11 pagesIMN1207 McGillActiveProtocolsapi-3714923No ratings yet
- Artigo OncoDocument14 pagesArtigo OncoCatarina LuísNo ratings yet
- Agreement On TradeDocument28 pagesAgreement On TradepraharshithaNo ratings yet
- Make Better Decisions - ShareDocument161 pagesMake Better Decisions - ShareDuyLENguyenHoangNo ratings yet
- Novartis DF 2006Document236 pagesNovartis DF 2006JORGENo ratings yet
- NovartisVsUnion LAB Team1 Pulkit Apoorva DebayudhDocument11 pagesNovartisVsUnion LAB Team1 Pulkit Apoorva DebayudhApoorva GoelNo ratings yet
- Chemistry of Anticancer Thiazole Compounds: Chawla Amit, Sheelmani, Arashdeep Singh, Chawla Payal, Dhawan R KDocument7 pagesChemistry of Anticancer Thiazole Compounds: Chawla Amit, Sheelmani, Arashdeep Singh, Chawla Payal, Dhawan R Kahmed magdyNo ratings yet
- Quiz 2 Name: Hafiz Abdul MananDocument2 pagesQuiz 2 Name: Hafiz Abdul Mananmanan abdulNo ratings yet
- Novartis AG V UOIDocument18 pagesNovartis AG V UOIRAJINI BOLLERA KUSHALAPPANo ratings yet
- Biopharmaceuticals Availability, Diffusion, Sustainability: Massimo Riccaboni University of Florence & CERM, RomeDocument28 pagesBiopharmaceuticals Availability, Diffusion, Sustainability: Massimo Riccaboni University of Florence & CERM, RomeNoor hossainNo ratings yet
- IPR ProjectDocument16 pagesIPR ProjectKanchan MehraNo ratings yet
- Cancer ChemotherapyDocument37 pagesCancer ChemotherapyRubyrose TagumNo ratings yet
- Imatinib - Drug Information PDFDocument20 pagesImatinib - Drug Information PDFabuzeid5No ratings yet
- Chronic Myelogenous Leukemia (CML) - Causes, Symptoms, TreatmentDocument10 pagesChronic Myelogenous Leukemia (CML) - Causes, Symptoms, Treatmentnurul auliaNo ratings yet
- Pruning The Evergreen Tree Section 3 (D) of The Indian Patents Act 1970Document29 pagesPruning The Evergreen Tree Section 3 (D) of The Indian Patents Act 1970Santosh PatiNo ratings yet
- Case Analysis Name of The Case Citation Date of Judgement Names of The Judge/s Provisions InvolvedDocument7 pagesCase Analysis Name of The Case Citation Date of Judgement Names of The Judge/s Provisions InvolvedyashNo ratings yet
- Mechanisms of Action of Commonly Used Drugs To Treat Cancer: Michael E. Trigg, MD, and Anne Flanigan-Minnick, PHDDocument13 pagesMechanisms of Action of Commonly Used Drugs To Treat Cancer: Michael E. Trigg, MD, and Anne Flanigan-Minnick, PHDFlorentina AdascaliteiNo ratings yet
- List of Anti-Cancer Drugs Aproved by The FDA - PharmaKnowDocument39 pagesList of Anti-Cancer Drugs Aproved by The FDA - PharmaKnowharsha2733No ratings yet