You might also like
- Pharmacology of Opioid AnalgesicsDocument64 pagesPharmacology of Opioid AnalgesicsDr.U.P.Rathnakar.MD.DIH.PGDHM100% (1)
- Ketamine For Opiate WithdrawalDocument3 pagesKetamine For Opiate WithdrawalroboNo ratings yet
- AlkaloidDocument33 pagesAlkaloidPoonam PandeyNo ratings yet
- Opoid AnalgesicsDocument29 pagesOpoid AnalgesicsShivsharan100% (1)
- Pharmacotherapeutics For Advanced Nursing Practice 2018Document790 pagesPharmacotherapeutics For Advanced Nursing Practice 2018Fadel AlJomaiaNo ratings yet
- Chapter - 31 - Opioid - Analgesics - Antagonist - PDF Filename UTF-8''Chapter 31 Opioid Analgesics & AntagonistDocument6 pagesChapter - 31 - Opioid - Analgesics - Antagonist - PDF Filename UTF-8''Chapter 31 Opioid Analgesics & AntagonistJoherNo ratings yet
- 4 Adrenergic AgonistsDocument46 pages4 Adrenergic Agonistsmatchees-gone rogueNo ratings yet
- AnalgesicDocument42 pagesAnalgesicVinayak SinghNo ratings yet
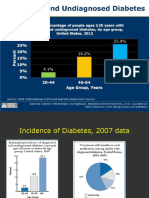
- Diabetes Mellitus in The US:: PrevalenceDocument48 pagesDiabetes Mellitus in The US:: PrevalenceokibreazyNo ratings yet
- Central Nervous System StimulantsDocument20 pagesCentral Nervous System StimulantsAn LoNo ratings yet
- Nervous System Diseases and DisordersDocument88 pagesNervous System Diseases and DisordersJape GarridoNo ratings yet
- Opioid Analgesics: Just in Time Training September 2006Document16 pagesOpioid Analgesics: Just in Time Training September 2006Yel CMNo ratings yet
- Opioid Analgesics: T. Binder Department of PharmacologyDocument36 pagesOpioid Analgesics: T. Binder Department of PharmacologywwwrgrobinNo ratings yet
- Morphine: Somniferum, Dried and Powdered - Opium PowderDocument4 pagesMorphine: Somniferum, Dried and Powdered - Opium PowderpharmbharathiNo ratings yet
- Hypnotics BarbituratesDocument34 pagesHypnotics BarbituratesIkram HamacheNo ratings yet
- SubstanceDocument11 pagesSubstancejrewingheroNo ratings yet
- Kuliah OpioidDocument50 pagesKuliah OpioidAndi RifkyNo ratings yet
- Tofisopam Medical 2014 REVISEDDocument97 pagesTofisopam Medical 2014 REVISEDdrashishnairNo ratings yet
- Antiseizure Drugs: By: Aim D. QuttenehDocument9 pagesAntiseizure Drugs: By: Aim D. QuttenehrimNo ratings yet
- PharmacogeneticsDocument3 pagesPharmacogeneticsPardeep Sony100% (1)
- Describe The Mechanism/s of Action, Types, Doses, Side Effects, Indications and Contraindications of Antidepressant DrugsDocument57 pagesDescribe The Mechanism/s of Action, Types, Doses, Side Effects, Indications and Contraindications of Antidepressant Drugssunil pandeyNo ratings yet
- Antimanic DrugsDocument22 pagesAntimanic DrugsMarlet N. Ortega100% (2)
- Classification of AlkaloidsDocument8 pagesClassification of AlkaloidsJenelle Jane Quilaneta100% (1)
- Anti-Anginal and Antiischemic Drugs: Dr. Pradeepa H D Clinical PharmacologistDocument25 pagesAnti-Anginal and Antiischemic Drugs: Dr. Pradeepa H D Clinical PharmacologistpradeephdNo ratings yet
- Anti Depressants FinalDocument61 pagesAnti Depressants FinalAuthor Nauman Shad100% (1)
- 04.04 Dermatologic PharmacologyDocument3 pages04.04 Dermatologic PharmacologyMaikka IlaganNo ratings yet
- Pharmacology of Ergot AlkaloidsDocument143 pagesPharmacology of Ergot AlkaloidsParveen Hullon100% (1)
- Documents Oral Morphine For Cancer Pain (Review) LDocument57 pagesDocuments Oral Morphine For Cancer Pain (Review) LWidayat WahyuNo ratings yet
- SARP (Skin Anesthesia Radiology Psychiatry) Review 2010Document4 pagesSARP (Skin Anesthesia Radiology Psychiatry) Review 2010QworldNo ratings yet
- General Pharmacology - Sources of Drugs and Routes of AdministrationDocument48 pagesGeneral Pharmacology - Sources of Drugs and Routes of AdministrationDhriti Brahma78% (9)
- Subject: Pharmaceutical Chemistry-II Chapter: Steroidal DrugsDocument19 pagesSubject: Pharmaceutical Chemistry-II Chapter: Steroidal DrugsRam PrajapatNo ratings yet
- Parkinson's DiseaseDocument65 pagesParkinson's DiseaseGerald Resubal OriñaNo ratings yet
- Clinical Alert A Quick Reference To Adverse Clinical EventsDocument272 pagesClinical Alert A Quick Reference To Adverse Clinical EventsOmasNers100% (1)
- Adjuvants DrugsDocument38 pagesAdjuvants DrugsIppank F SjNo ratings yet
- K10 (A1) - 2015pharmacotherapy For ParkinsonDocument41 pagesK10 (A1) - 2015pharmacotherapy For Parkinsonali100% (1)
- Antimanic DrugsDocument9 pagesAntimanic DrugscradletalkNo ratings yet
- Drug Receptor TheoryDocument9 pagesDrug Receptor TheoryFish YanNo ratings yet
- Antiobesity DrugsDocument5 pagesAntiobesity DrugsArjun MishraNo ratings yet
- Module # 5 Pharmacology NursingDocument45 pagesModule # 5 Pharmacology Nursingannyeong_123No ratings yet
- 13-Opioids Lecture 1Document41 pages13-Opioids Lecture 1api-343631539100% (1)
- Natural Product Chemistry For Drug DiscoveryDocument458 pagesNatural Product Chemistry For Drug DiscoveryUğur ÇiğdemNo ratings yet
- Adrenergic AgentsDocument57 pagesAdrenergic AgentsAn Lo100% (1)
- Acid Base BalanceDocument28 pagesAcid Base BalanceAnis BonitaNo ratings yet
- Principles of Opioid Management: Symptom GuidelinesDocument45 pagesPrinciples of Opioid Management: Symptom GuidelinesTheresia Avila KurniaNo ratings yet
- Opioid Peptides: Biology, Chemistry, and Genetics: The Peptides: Analysis, Synthesis, Biology, Vol. 6From EverandOpioid Peptides: Biology, Chemistry, and Genetics: The Peptides: Analysis, Synthesis, Biology, Vol. 6No ratings yet
- Substance Disorder 2020Document59 pagesSubstance Disorder 2020dennyzpsNo ratings yet
- Revellionz'19 - Second Year Question BankDocument114 pagesRevellionz'19 - Second Year Question BankRamNo ratings yet
- Opioid Analgesics: Dr. D. K. Brahma Department of Pharmacology, NEIGRIHMS Shillong, Meghalaya, ImdiaDocument48 pagesOpioid Analgesics: Dr. D. K. Brahma Department of Pharmacology, NEIGRIHMS Shillong, Meghalaya, ImdiaAmiraBenhammou100% (1)
- Drug MetabolismDocument27 pagesDrug MetabolismApurba Sarker ApuNo ratings yet
- Cell ReceptorsDocument10 pagesCell ReceptorsMaria PavalNo ratings yet
- Drug Interactions: DR - Omer A.A.S. Rikaby BSC Pharmacy MSC Clinical Pharmacy MbaDocument40 pagesDrug Interactions: DR - Omer A.A.S. Rikaby BSC Pharmacy MSC Clinical Pharmacy MbaAbdalla Kazzam100% (1)
- Sedative-Hypnotics (SeH) and AnxiolyticsDocument101 pagesSedative-Hypnotics (SeH) and Anxiolyticsmatchees-gone rogueNo ratings yet
- PharmacogeneticsDocument30 pagesPharmacogeneticsShailendra Sk100% (3)
- Resources For Medication Safety Lecture - ViswanathanDocument59 pagesResources For Medication Safety Lecture - Viswanathanapi-669020994No ratings yet
- AntipsychoticsDocument51 pagesAntipsychoticsShailja SharmaNo ratings yet
- Antiseizure Drugs - Mechanism of Action,... Cology, and Adverse Effects - UpToDateDocument82 pagesAntiseizure Drugs - Mechanism of Action,... Cology, and Adverse Effects - UpToDatecapt_zoeNo ratings yet
- Common Cardiac Related MedicationsDocument18 pagesCommon Cardiac Related MedicationsTracy100% (2)
- Apollo Prescription Flu VaccineDocument2 pagesApollo Prescription Flu VaccineSurajit BanerjeeNo ratings yet
- VRD Bortezomib Lenalidomide Dexamethasone Multiple Myeloma Protocol V1.0Document9 pagesVRD Bortezomib Lenalidomide Dexamethasone Multiple Myeloma Protocol V1.0gini erwantiNo ratings yet
- Drug Study #4Document7 pagesDrug Study #4Sarah Kaye BañoNo ratings yet
- DrugsDocument10 pagesDrugsEphraim Remann D. GarciaNo ratings yet
- Pedia Notes Compilation MKDoseDocument18 pagesPedia Notes Compilation MKDosemefav7778520No ratings yet
- Harga Prolanis (Data Dari Apotek Online)Document74 pagesHarga Prolanis (Data Dari Apotek Online)Orin Tri WulanNo ratings yet
- Reg ApotekDocument184 pagesReg ApotekjuwanNo ratings yet
- Daftar Obat KemoterapiDocument3 pagesDaftar Obat KemoterapiCharles Nong MakingNo ratings yet
- Benzodiazepine and Z Hypnotic Prescribing GuidanceDocument11 pagesBenzodiazepine and Z Hypnotic Prescribing GuidanceKru PrimeNo ratings yet
- Intro-Generic vs. Brand Medicines-An OverviewDocument8 pagesIntro-Generic vs. Brand Medicines-An OverviewManikanta GupthaNo ratings yet
- Hydroxyzine GuidelinesDocument13 pagesHydroxyzine GuidelinesMahim JainNo ratings yet
- Anestesi LokalDocument25 pagesAnestesi LokalAndi Upik FathurNo ratings yet
- Advanced Marketing Strategy: ZantacDocument6 pagesAdvanced Marketing Strategy: ZantacAALOK SINGLANo ratings yet
- listVetMeds PDFDocument26 pageslistVetMeds PDFsanathNo ratings yet
- Dispensing of Antibiotics Without Prescription Among The Community Pharmacies in Rupandehi District of NepalDocument72 pagesDispensing of Antibiotics Without Prescription Among The Community Pharmacies in Rupandehi District of NepalVictorious AtnapNo ratings yet
- ADHD Medication Conversion ChartDocument4 pagesADHD Medication Conversion ChartThaíse FontanaNo ratings yet
- Nasal Spray Condensed - HOW TO MAKE NASAL SPRAY FOR DRVGSDocument3 pagesNasal Spray Condensed - HOW TO MAKE NASAL SPRAY FOR DRVGSInés Grande BarrasNo ratings yet
- Design Features of Community PharmacyDocument5 pagesDesign Features of Community PharmacyIti ChauhanNo ratings yet
- Drug Administered Through The Following Route Is Most Likely To Be Subjected To First-Pass MetabolismDocument7 pagesDrug Administered Through The Following Route Is Most Likely To Be Subjected To First-Pass MetabolismpradeephdNo ratings yet
- Prescriptions AuditDocument22 pagesPrescriptions Auditdedicated atrishNo ratings yet
- Atypical Antipsychotics.Document9 pagesAtypical Antipsychotics.Said PerezNo ratings yet
- KFK200 PD-2 HT22Document40 pagesKFK200 PD-2 HT22Lihle MNo ratings yet
- نسخة 264889196 Tramadol Drug StudyDocument1 pageنسخة 264889196 Tramadol Drug StudyMeteab AlzhiryNo ratings yet
- Update Classification of Quinolone AntibioticDocument15 pagesUpdate Classification of Quinolone Antibioticsuvasish0068372No ratings yet
- Benzocaine Synthesis PDFDocument2 pagesBenzocaine Synthesis PDFLive FlightsNo ratings yet